Genome Annotations¶
- Learning Objective
- You will learn how to work with annotations in SeqAn. After this tutorial, you will be able to write your own programs using annotations and analyzing them. You will be ready to continue with the Fragment Store Tutorial, e.g. if you want to combine your annotations with information from alignments.
- Difficulty
- Average
- Duration
- 1 h
- Prerequisites
- Sequences, Iterators
This tutorial will present SeqAn’s efficient and easy-to-use data structures to work with annotations. They allow to annotate genome regions with features like ‘gene’, ‘mRNA’, ‘exon’, ‘intron’ and if required with custom features. We will give you an understanding of how to load annotations from a GFF or GTF file, store them in efficient data structures, as well as how to traverse and access these information.
AnnotationStore as Part of the FragmentStore¶
This section will give you a short introduction to data structures relevant for working with annotations.
In SeqAn, annotations are stored in the so-called annotationStore, which is part of the FragmentStore. The annotationStore can only be used together with the FragmentStore, because the latter stores additional information, e.g. the contig names or sequences. The FragmentStore is a data structure specifically designed for read mapping, genome assembly or gene annotation.
The FragmentStore can be seen as a database, where each table (called “store”) is implemented as a String. Each row of the table corresponds to an element in the string. The position of each element in the string implicitly represents the Id of such element in the table. All such strings are members of the class FragmentStore, are always present and empty if unused. For example, the member contigStore is a string of elements, each one containing among others a contig sequence.
For detailed information about the FragmentStore read the Fragment Store Tutorial.
Accordingly, the annotationStore is a String, where each element represents one annotation. Each element holds the necessary information, e.g. beginPos, endPos, parentId etc., as data members.
AnnotationStore¶
In this section you will learn how to work with the annotationStore itself.
Annotations are represented hierarchically by a tree having at least a root node.
A typical annotation tree looks as follows.
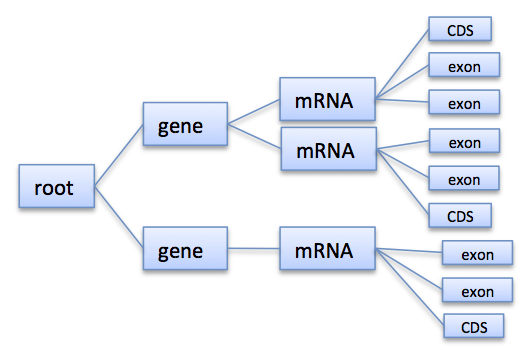
Annotation tree example
The following entity-relationship diagram shows the tables holding store annotations, their relationships and cardinalities.
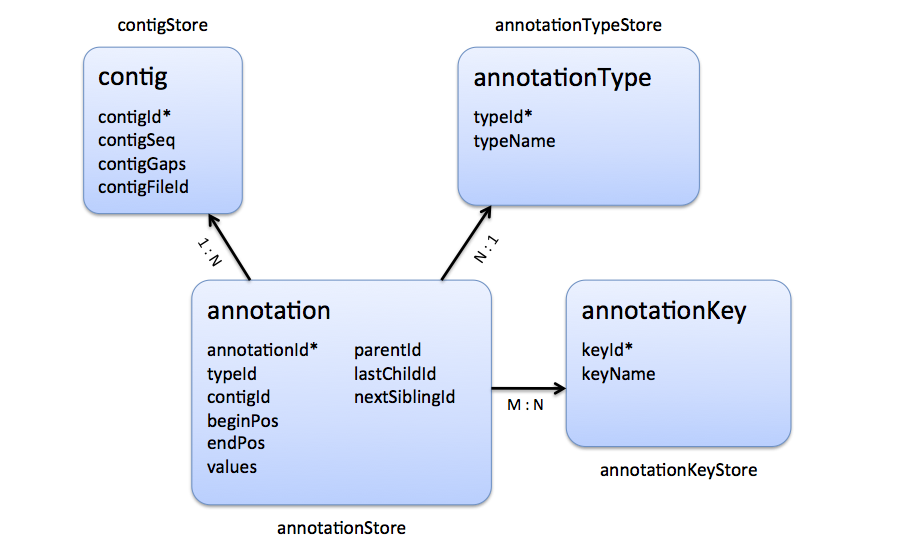
Stores involved in gene annotation
The instantiation of an annotationStore happens implicitly with the instantiation of a FragmentStore. Therefore we simply type:
FragmentStore<> store;
Loading an Annotation File¶
Before we deal with the actual annotation tree, we will first describe how you can easily load annotations from a GFF or GTF file into the FragmentStore.
An annotation file can be read from an open input stream with the function read. A tag specifies if we want to read a GFF, GTF or UCSC file. The following example shows how to read an GTF file:
// Open input stream from the current directory
std::ifstream file("example.gtf", std::ios_base::in | std::ios_base::binary);
// Read annotations from the GTF file
read(file, store, Gtf());
The GFF-reader is also able to detect and read GTF files. The UCSC Genome Browser uses two seperate files, the kownGene.txt and knownIsoforms.txt. They must be read by two consecutive calls of read (first knownGene.txt then knownIsoforms.txt).
Tip
An annotation can be loaded without loading the corresponding contigs.
In that case empty contigs are created in the contigStore with names given in the annonation. A subsequent call of loadContigs would load the sequences of these contigs, if they have the same identifier in the contig file.
Traversing the Annotation Tree¶
This section will illustrate how to use iterators to traverse the annotation tree.
The annotation tree can be traversed and accessed with the AnnotationTree Iterator. Again we use the metafunction dox:ContainerConcept#Iterator Iterator to determine the appropriate iterator type for our container. A new AnnotationTree iterator can be obtained by calling begin with a reference to the FragmentStore and the AnnotationTree tag:
Iterator<FragmentStore<>, AnnotationTree<> >::Type it;
it = begin(store, AnnotationTree<>());
The AnnotationTree iterator starts at the root node and can be moved to adjacent tree nodes with the functions goDown, goUp, and goRight. These functions return a boolean value that indicates whether the iterator could be moved. The functions isLeaf, isRoot, isLastChild return the same boolean without moving the iterator. With goRoot or goTo the iterator can be moved to the root node or an arbitrary node given its annotationId. If the iterator should not be moved but a new iterator at an adjacent node is required, the functions nodeDown, nodeUp, nodeRight can be used.
// Move the iterator down to a leaf
while (goDown(it));
// Create a new iterator and if possible move it to the right sibling of the first iterator
Iterator<FragmentStore<>, AnnotationTree<> >::Type it2;
if (isLastChild(it))
it2 = nodeRight(it);
The AnnotationTree iterator supports a preorder DFS traversal and therefore can also be used in typical begin-end loops with the functions goBegin (== goRoot), goEnd, goNext, atBegin, atEnd. During a preorder DFS, the descent into subtree can be skipped by goNextRight, or goNextUp which proceeds with the next sibling or returns to the parent node and proceeds with the next node in preorder DFS.
// Move the iterator back to the beginning
goBegin(it);
// Iterate over the nodes in preorder DFS while the end is not reached and
// output if the current node is a leaf
for (goBegin(it); atEnd(it); goNext(it))
{
if (isLeaf(it))
std::cout << " current node is leaf" << std::endl;
}
Asignment 1¶
- Type
- Review
- Objective
Copy the code below, which loads the annotations from a given GTF file into the FragmentStore and initializes an iterator on the AnnotationTree. Download the GTF file assignment_annotations.gtf, whose annotations build an AnnotationTree of the typical structure with gene, mRNA and exon level. Adjust the code to go down to the exon level and iteratate over all children of the first mRNA and count them. Print the result.
Click more... to see the code.
#include <fstream> #include <iostream> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/store.h> using namespace seqan; int main() { FragmentStore<> store; std::ifstream file("assignment_annotations.gtf", std::ios_base::in | std::ios_base::binary); read(file, store, Gtf()); // Create AnnotationTree iterator Iterator<FragmentStore<>, AnnotationTree<> >::Type it; it = begin(store, AnnotationTree<>()); // Move iterator one node down goDown(it); return 0; }
- Hints
- In the given data the left-most leaf is a child of mRNA and has siblings. You can use the function goRight to traverse over all siblings.
- Solution
Click more... to see one possible solution.
#include <fstream> #include <iostream> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/store.h> using namespace seqan; int main() { FragmentStore<> store; std::ifstream file("assignment_annotations.gtf", std::ios_base::in | std::ios_base::binary); read(file, store, Gtf()); // Create AnnotationTree iterator Iterator<FragmentStore<>, AnnotationTree<> >::Type it; it = begin(store, AnnotationTree<>()); unsigned count = 0; // Go down to the first leaf (first child of the first mRNA) while (goDown(it)) ; ++count; // Iterate over all siblings and count while (goRight(it)) ++count; std::cout << "No. of children of the first mRNA: " << count << std::endl; return 0; }
No. of children of the first mRNA: 9
Assignment 2¶
- Type
- Review
- Objective
- Reuse the code and the GTF file from above. Instead of counting only the children of the first mRNA adjust the code to count the children for each given mRNA. Print the results.
- Hints
- After you reached the last child of the first mRNA you can use the functions goNext and goDown to traverse to the next leaf.
- Solution
Click more... to see one possible solution.
#include <fstream> #include <iostream> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/store.h> using namespace seqan; int main() { FragmentStore<> store; std::ifstream file("assignment_annotations.gtf", std::ios_base::in | std::ios_base::binary); read(file, store, Gtf()); // Iterate over all leafs, count and print the result Iterator<FragmentStore<>, AnnotationTree<> >::Type it; it = begin(store, AnnotationTree<>()); unsigned count = 0; std::cout << "Number of children for each mRNA: " << std::endl; // Go down to the first leaf (first child of the first mRNA) while (goDown(it)) ; while (!atEnd(it)){ ++count; // Iterate over all siblings and count while (goRight(it)) ++count; std::cout << count << std::endl; count = 0; // Jump to the next mRNA or gene, go down to its first leaf and count it if (!atEnd(it)) { goNext(it); if (!atEnd(it)) while(goDown(it)) ; } } return 0; }
9 2 2
Accessing the Annotation Tree¶
Let us now have a closer look how to access the information stored in the different stores representing the annotation tree.
To access or modify the node an iterator points at, the iterator returns the node’s annotationId by the value function (== operator*). With the annotationId the corresponding entry in the annotationStore could be modified manually or by using convenience functions. The function getAnnotation returns a reference to the corresponding entry in the annotationStore. getName and setName can be used to retrieve or change the identifier of the annotation element. As some annotation file formats don’t give every annotation a name, the function getUniqueName returns the name if non-empty or generates one using the type and id. The name of the parent node in the tree can be determined with getParentName. The name of the annotation type, e.g. ‘mRNA’ or ‘exon’, can be determined and modified with getType and setType.
Assume we have loaded the file example.gtf with the following content to the FragmentStore store and instantiated the iterator it of the corresponding annotation tree.
chr1 MySource exon 150 200 . + . gene_id "381.000"; transcript_id "381.000.1";
chr1 MySource exon 300 401 . + . gene_id "381.000"; transcript_id "381.000.1";
chr1 MySource CDS 380 401 . + 0 gene_id "381.000"; transcript_id "381.000.1";
chr1 MySource exon 160 210 . + . gene_id "381.000"; transcript_id "381.000.2";
We now want to iterate to the first exon and output a few information:
// Move the iterator to the begin of the annotation tree
it = begin(store, AnnotationTree<>());
// Go down to exon level
while (goDown(it)) ;
std::cout << "type: " << getType(it) << std::endl;
std::cout << "id: " << value(it) << std::endl;
std::cout << "begin position: " << getAnnotation(it).beginPos << std::endl;
For our example the output would be:
type: exon
id: 3
begin position: 149
An annotation can not only refer to a region of a contig but also contain additional information given as key-value pairs. The value of a key can be retrieved or set by getValueByKey and assignValueByKey. The values of a node can be cleared with clearValues.
A new node can be created as first child, last child, or right sibling of the current node with createLeftChild, createRightChild, or createSibling. All three functions return an iterator to the newly created node.
Iterator<FragmentStore<>, AnnotationTree<> >::Type it2;
// Create a right sibling of the current node and return an iterator to this new node
it2 = createSibling(it);
The following list summarizes the functions provided by the AnnotationTree iterator.
- getAnnotation, value
- Return annotation object/id of current node
- getName, setName, getType, setType
- Access name or type of current annotation object
- getParentName
- Access parent name of current annotation object
- clearValue, getValueByKey, assignValueByKey
- Access associated values
- goBegin, goEnd, atBegin, atEnd
- Go to or test for begin/end of DFS traversal
- goNext, goNextRight, goNextUp
- go next, skip subtree or siblings during DFS traversal
- goRoot, goUp, goDown, goRight
- Navigate through annotation tree
- createLeftChild, createRightChild, createSibling
- Create new annotation nodes
- isRoot, isLeaf
- Test for root/leaf node
Assignment 3¶
- Type
- Application
- Objective
Again use the given GTF file assignment_annotations.gtf and create an iterator on the annotation tree. Now iterate to the first node of type “exon” and output the following features:
- type
- begin position
- end position
- its Id
- the Id of its parent
- the name of its parent
- Solution
Click more... to see one possible solution.
#include <fstream> #include <iostream> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/store.h> using namespace seqan; int main() { FragmentStore<> store; std::ifstream file("assignment_annotations.gtf", std::ios_base::in | std::ios_base::binary); read(file, store, Gtf()); // Create iterator Iterator<FragmentStore<>, AnnotationTree<> >::Type it; it = begin(store, AnnotationTree<>()); // Iterate to the first annotation of type "exon" while (!atEnd(it) && getType(it) != "exon") goNext(it); // Output: std::cout << " type: " << getType(it) << std::endl; std::cout << " begin position: " << getAnnotation(it).beginPos << std::endl; std::cout << " end position: " << getAnnotation(it).endPos << std::endl; std::cout << " id: " << value(it) << std::endl; std::cout << " parent id: " << getAnnotation(it).parentId << std::endl; std::cout << " parent name: " << getParentName(it) << std::endl; return 0; }
type: exon begin position: 149 end position: 200 id: 3 parent id: 2 parent name: 381.000.1
Assignment 4¶
- Objective
Write a small statistic tool to analyse a given set of annotations.
- Load the annotations given in the GTF file assignment_annotations.gtf.
- Output the average number of mRNAs for genes.
- Output the average number of exons for mRNAs.
- Additionally output the average exon length.
- Test your program also on large data, e.g. the annotation of the mouse genome [raw-attachment:Mus_musculus.NCBIM37.61.gtf.zip:wiki:Tutorial/SimpleRnaSeq Mus_musculus.NCBIM37.61.gtf.zip] (don’t forget to unzip first).
- Solution
Click more... to see one possible solution.
#include <fstream> #include <iostream> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/store.h> using namespace seqan; int main() { FragmentStore<> store; std::ifstream file("assignment_annotations.gtf", std::ios_base::in | std::ios_base::binary); read(file, store, Gtf()); // Create iterator Iterator<FragmentStore<>, AnnotationTree<> >::Type it; it = begin(store, AnnotationTree<>()); unsigned countGenes = 0; unsigned countmRNAs = 0; unsigned countExons = 0; unsigned length = 0; // Iterate over annotation tree and count different elements and compute exon lengths while (!atEnd(it)){ if (getType(it) == "gene") ++countGenes; else if (getType(it) == "mRNA") ++countmRNAs; else if (getType(it) == "exon") { ++countExons; length += abs((int)getAnnotation(it).endPos - (int)getAnnotation(it).beginPos); } goNext(it); } // Ouput some stats: std::cout << "Average number of mRNAs for genes: " << (float)countmRNAs/(float)countGenes << std::endl; std::cout << "Average number of exons for mRNAs: " << (float)countExons/(float)countmRNAs << std::endl; std::cout << "Average length of exons: " << (float)length/(float)countExons << std::endl; return 0; }
Average number of mRNAs for genes: 1.5 Average number of exons for mRNAs: 3 Average length of exons: 95.5556
Write an Annotation File¶
To write an annotation to an open output stream use the function write and specify the file format with a tag Gff() or Gtf().
// Open output stream
std::ofstream fileOut("example_out.gtf", std::ios_base::out | std::ios_base::binary);
// Write annotations to GTF file
write(fileOut, store, Gtf());