Mini Bowtie¶
- Learning Objective
- You will be able to write read mappers using SeqAn.
- Difficulty
- Hard
- Duration
- 2h
- Prerequisites
- Indices, Index Iterators, Fragment Store
Mini-Bowtie is a very basic read aligner that is inspired by the well known Bowtie program [LTP+09]. It serves as an example to show that you can write sophisticated programs with SeqAn using few lines of code.
Background¶
Bowtie is based on the FM index [FM01] which uses the Burrows-Wheeler transform (BWT) to search for patterns in a text in \(\mathcal{O}(m)\) time with m being the pattern length. Furthermore the index only consumes minute space because it can be compressed using favorable features of the BWT.
Since this tutorial serves as an example of SeqAn’s functionality, we will not completely re-implement Bowtie. Instead we will concentrate on providing a simple yet functional read mapper. We will start by reading the reference sequence and reads, then search approximately each read in the reference allowing at most one mismatch, and finally write the result into an informative SAM file.
We do make use of the FM index, but we use only one key feature of Bowtie, namely a two phase search. Such two phase search speeds up approximate search on the FM index but requires two indices, i.e. one of the forward and one of the backward reference sequence. Instead of searching each read in one phase, allowing one mismatch in every possible position, we search it twice, but in two phases having some restrictions. We first search each read in the forward index by allowing one mismatch only in the second half of the read. Then we reverse each read and search it in the backward index, again by allowing one mismatch only in the second half of the read. The advantage of a two phase search will become clear in the following.
Tip
The FM index implicitly represent a prefix trie of a text. Searching on the FM index is performed backwards, starting with the last character of the pattern and working its way to the first.
In the first search phase we search backwards the right half of the read without mismatches. If we succeed, in the second phase we continue backwards allowing one mismatch in the left half. The whole procedure is based on a depth-first traversal on the prefix trie of the reference. Trie’s edges are labeled with single characters and each path following some edges spells a substring of the reference. Thus, first we start in the root node and follow the path spelling the right half of the read. If we succeed, we continue in the subtree rooted in the previously reached node by following each possible path that spells the left half of the read with one substitution.
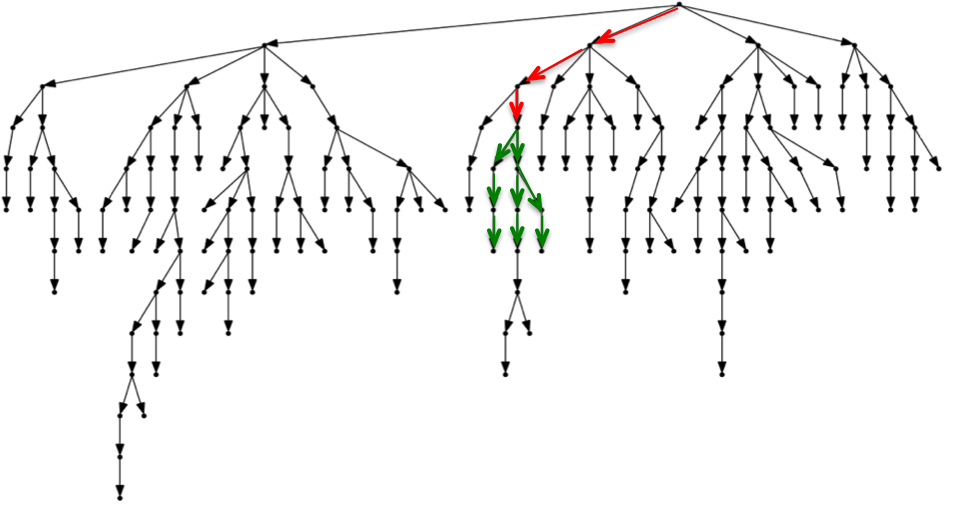
The red arrows indicate the exact search of the right half of the read, the green arrows the approximate search of the left half.
From the figure above you can see the advantage of using a two phase approach. By starting from the root node and performing exact search, we follow only one path and avoid visiting the top of the trie. Approximate search is more involved, since we have to try every path until we find a second mismatch, but it is limited to a small subtree.
So far we have found all locations of a read having at most one mismatch in its left half. In order to find also all locations having at most one mismatch in the right half, we simply reverse the reference and the read and repeat the procedure from above. Note that we only reverse but do not complement the reference or the reads.
Assignment 1¶
- Type
- Application
- Objective
Copy the code below and rearrange the rows such that they give a functional order.
// ========================================================================== // mini_bowtie // ========================================================================== // Copyright (c) 2006-2012, Knut Reinert, FU Berlin // All rights reserved. #include <iostream> #include <seqan/basic.h> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/index.h> #include <seqan/store.h> using namespace seqan; void search() {}; int main(int argc, char \*argv[]) { // type definitions typedef String<Dna5> TString; typedef StringSet<TString> TStringSet; typedef Index<StringSet<TString>, FMIndex<> > TIndex; typedef Iterator<TIndex, TopDown<ParentLinks<> > >::Type TIter; // reversing the sequences for backward search // backward search // reading the command line arguments // declaration and initialization of the fragment store // forward search // combining the contigs of the reference into one string set appendValue(text, fragStore.contigStore[i].seq); std::cerr << "Invalid number of arguments." << std::endl << "USAGE: mini_bowtie GENOME.fasta READS.fasta OUT.sam" << std::endl; } if (argc < 3) { if (loadContigs(fragStore, argv[1])) return 1; if (loadReads(fragStore, argv[2])) return 1; clear(fmIndex); clear(fmIndex); StringSet<TString> text; for (unsigned i = 0; i < length(fragStore.contigStore); ++i) fmIndex = TIndex(text); TIndex fmIndex(text); TIter it(fmIndex); search(); search(); clear(it); clear(it); reverse(text); reverse(fragStore.readSeqStore); it = TIter(fmIndex); FragmentStore<> fragStore; return 0; return 1; }- Hint
- We make use of the FragmentStore. While we can access the pattern/reads as if using a StringSet, we need to create a StringSet of the contigs, because in the FragmentStore the contigs are not stored in a StringSet.
- Hint
The correct order of the comments is:
// reading the command line arguments // declaration and initialization of the fragment store // combining the contigs of the reference into one string set // forward search // reversing the sequences for backward search // backward search
- Solution
// ========================================================================== // mini_bowtie // ========================================================================== #include <iostream> #include <seqan/basic.h> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/index.h> #include <seqan/store.h> using namespace seqan; void search() {} int main(int argc, char *argv[]) { // type definitions typedef String<Dna5> TString; typedef StringSet<TString> TStringSet; typedef Index<StringSet<TString>, FMIndex<> > TIndex; typedef Iterator<TIndex, TopDown<ParentLinks<> > >::Type TIter; // reading the command line arguments if (argc < 3) { std::cerr << "Invalid number of arguments." << std::endl << "USAGE: minimapper GENOME.fasta READS.fasta OUT.sam" << std::endl; return 1; } // declaration and initialization of the fragment store FragmentStore<> fragStore; if (!loadContigs(fragStore, argv[1])) return 1; if (!loadReads(fragStore, argv[2])) return 1; StringSet<TString> text; for (unsigned i = 0; i < length(fragStore.contigStore); ++i) appendValue(text, fragStore.contigStore[i].seq); TIndex fmIndex(text); TIter it(fmIndex); search(); clear(fmIndex); clear(it); reverse(text); reverse(fragStore.readSeqStore); fmIndex = TIndex(text); it = TIter(fmIndex); search(); clear(fmIndex); clear(it); reverse(text); reverse(fragStore.readSeqStore); clear(fmIndex); clear(it); return 0; }
Now that we have the backbone of our program we can start to implement the fundamental part, the search routine. The search function requires two input arguments, namely the iterator used to traverse the FM index of the reference sequence and the string set containing the reads.
The search function iterates over the reads and searches them in the already mentioned two phase fashion. In the first phase the right half of the pattern is searched exactly. The second phase is more involved and will be discussed after the second assignment.
Assignment 2¶
- Type
- Application
- Objective
- Expand the solution to the last assignment by implementing the backbone of the search routine. The backbone should consist of function definition, an outer loop traversing the pattern (using a standard iterator) and the first step of the search, namely the exact search of the right pattern half.
- Hint
The template function header could look like this:
template <typename TIter, typename TStringSet> void search(TIter & it, TStringSet const & pattern)
- Hint
You can obtain the correct iterator type using the metafunction Iterator.
typedef typename Iterator<TStringSet const, Standard>::Type TPatternIter;
- Solution
// ========================================================================== // mini_bowtie // ========================================================================== #include <iostream> #include <seqan/basic.h> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/index.h> #include <seqan/store.h> using namespace seqan; template <typename TIter, typename TStringSet> void search(TIter & it, TStringSet const & pattern) { typedef typename Iterator<TStringSet const, Standard>::Type TPatternIter; for (TPatternIter patternIt = begin(pattern, Standard()); patternIt != end(pattern, Standard()); ++patternIt) { unsigned startApproxSearch = length(value(patternIt)) / 2; goDown(it, infix(value(patternIt), 0, startApproxSearch - 1)); goRoot(it); } } int main(int argc, char *argv[]) { typedef String<Dna5> TString; typedef StringSet<String<Dna5> > TStringSet; typedef Index<StringSet<TString>, FMIndex<> > TIndex; typedef Iterator<TIndex, TopDown<ParentLinks<> > >::Type TIter; /*String<Dna> text = "ACGTACGT"; Index<String<Dna>, FMIndex<> > index(text); Finder<Index<String<Dna> > > finder(index); find(finder, "AC"); std::cerr << position("AC") << std::endl; */ // reading the command line arguments if (argc < 3) { std::cerr << "Invalid number of arguments." << std::endl << "USAGE: minimapper GENOME.fasta READS.fasta OUT.sam" << std::endl; return 1; } // declaration and initialization of the fragment store FragmentStore<> fragStore; if (!loadContigs(fragStore, argv[1])) return 1; if (!loadReads(fragStore, argv[2])) return 1; // combining the contigs of the reference into one string set StringSet<TString> text; for (unsigned i = 0; i < length(fragStore.contigStore); ++i) appendValue(text, fragStore.contigStore[i].seq); // forward search reverse(text); TIndex fmIndex(text); TIter it(fmIndex); search(it, fragStore.readSeqStore); clear(fmIndex); clear(it); // reversing the sequences for backward search reverse(text); reverse(fragStore.readSeqStore); // backward search fmIndex = TIndex(text); it = TIter(fmIndex); search(it, fragStore.readSeqStore); clear(fmIndex); clear(it); return 0; }
At this point we need to implement the most critical part of the our program, which is the second step of the search phase. In our case this step works as follows:
Assume that we have already found a path in the trie representing the pattern from position \(i\) to \((m - 1)\) with m being the pattern length and \(i < m / 2\). Then we substitute the character of the pattern at position \(i - 1\) with every character of the alphabet and search for the remaining characters exact. We repeat those two steps until we processed every character of the pattern.
The corresponding pseudo code could look like this:
unsigned startApproxSearch = length(pattern) / 2;
for i = startApproxSearch to 0
{
for all c in the aphabet
{
if (goDown(it, c))
{
if (goDown(it, pattern[0..i - 1]))
{
HIT
}
goBack to last start
}
}
goDown correct character
}
Assignment 3¶
- Type
- Application
- Objective
- Include the pseudo code from above into the search function.
- Hint
- Make a copy of the iterator before following the path of the substituted character. Doing so saves time and keeps the code simple because you do not need to use goUp.
- Hint
- goDown returns a boolean indicating if a path exists or not. In addition, you do not need to go through the steps of the pseudo code if the second pattern half was not found!
- Hint
- Solution
// ========================================================================== // mini_bowtie // ========================================================================== #include <iostream> #include <seqan/basic.h> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/index.h> #include <seqan/store.h> using namespace seqan; template <typename TIter, typename TStringSet> void search(TIter & it, TStringSet const & pattern) { typedef typename Iterator<TStringSet const, Standard>::Type TPatternIter; for (TPatternIter patternIt = begin(pattern, Standard()); patternIt != end(pattern, Standard()); ++patternIt) { unsigned startApproxSearch = length(value(patternIt)) / 2; if (goDown(it, infix(value(patternIt), 0, startApproxSearch - 1))) { for (unsigned i = startApproxSearch; ; ++i) { for (Dna5 c = MinValue<Dna>::VALUE; c < valueSize<Dna>(); ++c) { TIter localIt = it; if (goDown(localIt, c)) { if (goDown(localIt, infix(value(patternIt), i + 1, length(value(patternIt))))) { // HIT } } } if (i == length(value(patternIt)) - 1 || !goDown(it, getValue(patternIt)[i])) { break; } } } goRoot(it); } } int main(int argc, char *argv[]) { typedef String<Dna5> TString; typedef StringSet<String<Dna5> > TStringSet; typedef Index<StringSet<TString>, FMIndex<> > TIndex; typedef Iterator<TIndex, TopDown<> >::Type TIter; // 0) Handle command line arguments. if (argc < 3) { std::cerr << "Invalid number of arguments." << std::endl << "USAGE: minimapper GENOME.fasta READS.fasta OUT.sam" << std::endl; return 1; } // 1) Load contigs and reads. FragmentStore<> fragStore; if (!loadContigs(fragStore, argv[1])) return 1; if (!loadReads(fragStore, argv[2])) return 1; StringSet<TString> text; for (unsigned i = 0; i < length(fragStore.contigStore); ++i) appendValue(text, fragStore.contigStore[i].seq); reverse(text); TIndex fmIndex(text); TIter it(fmIndex); search(it, fragStore.readSeqStore); clear(fmIndex); clear(it); reverse(text); reverse(fragStore.readSeqStore); fmIndex = TIndex(text); it = TIter(fmIndex); search(it, fragStore.readSeqStore); clear(fmIndex); clear(it); return 0; }
So far so good. But there is a slight mistake. While substituting we also substitute the character in the pattern with itself. Therefore we find locations of exact matches multiple times.
Assignment 4¶
- Type
- Application
- Objective
- Adjust the code to cope with the problem mentioned above.
- Solution
// ========================================================================== // mini_bowtie // ========================================================================== #include <iostream> #include <seqan/basic.h> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/index.h> #include <seqan/store.h> using namespace seqan; template <typename TIter, typename TStringSet> void search(TIter & it, TStringSet const & pattern) { typedef typename Iterator<TStringSet const, Standard>::Type TPatternIter; for (TPatternIter patternIt = begin(pattern, Standard()); patternIt != end(pattern, Standard()); ++patternIt) { // exact search on pattern half unsigned startApproxSearch = length(value(patternIt)) / 2; if (goDown(it, infix(value(patternIt), 0, startApproxSearch - 1))) { for (unsigned i = startApproxSearch; ; ++i) { Dna character = getValue(patternIt)[i]; for (Dna5 c = MinValue<Dna>::VALUE; c < valueSize<Dna>(); ++c) { if (c != character) { TIter localIt = it; if (goDown(localIt, c)) { if (goDown(localIt, infix(value(patternIt), i + 1, length(value(patternIt))))) { //HIT } } } } if (!goDown(it, character)) break; else if (i == length(value(patternIt)) - 1) { // HIT break; } } } goRoot(it); } } int main(int argc, char *argv[]) { typedef String<Dna5> TString; typedef StringSet<String<Dna5> > TStringSet; typedef Index<StringSet<TString>, FMIndex<> > TIndex; typedef Iterator<TIndex, TopDown<> >::Type TIter; // 0) Handle command line arguments. if (argc < 3) { std::cerr << "Invalid number of arguments." << std::endl << "USAGE: minimapper GENOME.fasta READS.fasta OUT.sam" << std::endl; return 1; } // 1) Load contigs and reads. FragmentStore<> fragStore; if (!loadContigs(fragStore, argv[1])) return 1; if (!loadReads(fragStore, argv[2])) return 1; StringSet<TString> text; for (unsigned i = 0; i < length(fragStore.contigStore); ++i) appendValue(text, fragStore.contigStore[i].seq); reverse(text); TIndex fmIndex(text); TIter it(fmIndex); search(it, fragStore.readSeqStore); clear(fmIndex); clear(it); reverse(text); reverse(fragStore.readSeqStore); fmIndex = TIndex(text); it = TIter(fmIndex); search(it, fragStore.readSeqStore); clear(fmIndex); clear(it); reverse(text); reverse(fragStore.readSeqStore); std::ofstream samFile(argv[3], std::ios_base::out); write(samFile, fragStore, Sam()); return 0; }
So this is already the fundamental part of our program. What’s left to do is to write the result into a SAM file. In order to do so, we make use of the FragmentStore. Everything we need to do is to fill the alignedReadStore which is a member of the FragmentStore. This is very easy, because we only need to append a new value of type AlignedReadStoreElement specifying the match id, the pattern id, the id of the contig, as well as the begin and end position of the match in the reference.
An addMatchToStore function could look like this:
template <typename TStore, typename TIter, typename TPatternIt>
void addMatchToStore(TStore & fragStore, TPatternIt const & patternIt, TIter const & localIt)
{
typedef FragmentStore<>::TAlignedReadStore TAlignedReadStore;
typedef Value<TAlignedReadStore>::Type TAlignedRead;
for (unsigned num = 0; num < countOccurrences(localIt); ++num)
{
unsigned pos = getOccurrences(localIt)[num].i2;
TAlignedRead match(length(fragStore.alignedReadStore), position(patternIt), getOccurrences(localIt)[num].i1 ,
pos, pos + length(value(patternIt)));
appendValue(fragStore.alignedReadStore, match);
}
}
One could think we are done now. Unfortunately we are not. There are two problems. Recall, that in the second search phase we reverse the text and pattern, therefore we messed up start and end positions in the original reference. Furthermore we found exact matches twice, once in the forward index and once in the reversed index.
Assignment 5¶
- Type
- Application
- Objective
Include the following two lines into your code:
struct ForwardTag {}; struct ReverseTag {}; and write a second ``addMatchToStore`` function that is called when we search in the reversed reference. In addition, make all necessary changes to the code such that exact matches are only added once.``- Hint
- The metafunction IsSameType can be used to determine whether two types are equal or not.
- Solution
// ========================================================================== // mini_bowtie // ========================================================================== #include <iostream> #include <seqan/basic.h> #include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/index.h> #include <seqan/store.h> using namespace seqan; struct ForwardTag {}; struct ReverseTag {}; template <typename TStore, typename TIter, typename TPatternIt> void addMatchToStore(TStore & fragStore, TPatternIt const & patternIt, TIter const & localIt, ForwardTag) { typedef FragmentStore<>::TAlignedReadStore TAlignedReadStore; typedef Value<TAlignedReadStore>::Type TAlignedRead; for (unsigned num = 0; num < countOccurrences(localIt); ++num) { unsigned pos = getOccurrences(localIt)[num].i2; TAlignedRead match(length(fragStore.alignedReadStore), position(patternIt), getOccurrences(localIt)[num].i1 , pos, pos + length(value(patternIt))); appendValue(fragStore.alignedReadStore, match); } } template <typename TStore, typename TIter, typename TPatternIt> void addMatchToStore(TStore & fragStore, TPatternIt const & patternIt, TIter const & localIt, ReverseTag) { typedef FragmentStore<>::TAlignedReadStore TAlignedReadStore; typedef Value<TAlignedReadStore>::Type TAlignedRead; for (unsigned num = 0; num < countOccurrences(localIt); ++num) { unsigned contigLength = length(fragStore.contigStore[getOccurrences(localIt)[num].i1].seq); unsigned pos = contigLength - getOccurrences(localIt)[num].i2 - length(value(patternIt)); TAlignedRead match(length(fragStore.alignedReadStore), position(patternIt), getOccurrences(localIt)[num].i1, pos, pos + length(value(patternIt))); appendValue(fragStore.alignedReadStore, match); } } template <typename TIter, typename TStringSet, typename TStore, typename DirectionTag> void search(TIter & it, TStringSet const & pattern, TStore & fragStore, DirectionTag /*tag*/) { typedef typename Iterator<TStringSet const, Standard>::Type TPatternIter; for (TPatternIter patternIt = begin(pattern, Standard()); patternIt != end(pattern, Standard()); ++patternIt) { // exact search on pattern half unsigned startApproxSearch = length(value(patternIt)) / 2; if (goDown(it, infix(value(patternIt), 0, startApproxSearch - 1))) { for (unsigned i = startApproxSearch; ; ++i) { Dna character = getValue(patternIt)[i]; for (Dna5 c = MinValue<Dna>::VALUE; c < valueSize<Dna>(); ++c) { if (c != character) { TIter localIt = it; if (goDown(localIt, c)) { if (goDown(localIt, infix(value(patternIt), i + 1, length(value(patternIt))))) { addMatchToStore(fragStore, patternIt, localIt, DirectionTag()); } } } } if (!goDown(it, character)) break; else if (i == length(value(patternIt)) - 1) { if(IsSameType<DirectionTag, ForwardTag>::VALUE) addMatchToStore(fragStore, patternIt, it, DirectionTag()); break; } } } goRoot(it); } } int main(int argc, char *argv[]) { typedef String<Dna5> TString; typedef StringSet<String<Dna5> > TStringSet; typedef Index<StringSet<TString>, FMIndex<> > TIndex; typedef Iterator<TIndex, TopDown<> >::Type TIter; // 0) Handle command line arguments. if (argc < 3) { std::cerr << "Invalid number of arguments." << std::endl << "USAGE: minimapper GENOME.fasta READS.fasta OUT.sam" << std::endl; return 1; } // 1) Load contigs and reads. FragmentStore<> fragStore; if (!loadContigs(fragStore, argv[1])) return 1; if (!loadReads(fragStore, argv[2])) return 1; StringSet<TString> text; for (unsigned i = 0; i < length(fragStore.contigStore); ++i) appendValue(text, fragStore.contigStore[i].seq); reverse(text); TIndex fmIndex(text); TIter it(fmIndex); search(it, fragStore.readSeqStore, fragStore, ForwardTag()); clear(fmIndex); clear(it); reverse(text); reverse(fragStore.readSeqStore); fmIndex = TIndex(text); it = TIter(fmIndex); search(it, fragStore.readSeqStore, fragStore, ReverseTag()); clear(fmIndex); clear(it); reverse(text); reverse(fragStore.readSeqStore); std::ofstream samFile(argv[3], std::ios_base::out); write(samFile, fragStore, Sam()); return 0; }
Done? Not quite.
We need to copy the following four lines of code into our code in order to write the correct result in the SAM file. Calling the reverse function is necessary because an alignment must be computed for every match to be written into the SAM file.
reverse(text);
reverse(fragStore.readSeqStore);
std::ofstream samFile(argv[3], std::ios_base::out);
write(samFile, fragStore, Sam());