Fragment Store¶
- Learning Objective
- You will learn about the SeqAn FragmentStore for handling fragments. The “fragments” are reads and the data structure is useful in the context of read mapping, genome assembly, and gene annotation. After completing this tutorial, you will be able to use the most relevant functionality of the FragmentStore class.
- Difficulty
- Advanced
- Duration
- 1 h
- Prerequisites
- The basic tutorials.
The FragmentStore is a data structure specifically designed for read mapping, genome assembly or gene annotation. These tasks typically require lots of data structures that are related to each other like:
- reads, mate-pairs, reference genome
- pairwise alignments
- genome annotation
The Fragment Store subsumes all these data structures in an easy to use interface. It represents a multiple alignment of millions of reads or mate-pairs against a reference genome consisting of multiple contigs. Additionally, regions of the reference genome can be annotated with features like ‘gene’, ‘mRNA’, ‘exon’, ‘intron’ or custom features. The Fragment Store supports I/O functions to read/write a read alignment in SAM or AMOS format and to read/write annotations in GFF or GTF format.
The Fragment Store can be compared with a database where each table (called “store”) is implemented as a String member of the FragmentStore class. The rows of each table (implemented as structs) are referred by their ids which are their positions in the string and not stored explicitly (marked with * in the Figures 2 and 5). The only exception is the alignedReadStore whose elements of type AlignedReadStoreElement contain an id-member as they may be rearranged in arbitrary order, e.g. by increasing genomic positions or by readId. Many stores have an associated name store to store element names. Each name store is a StringSet that stores the element name at the position of its id. All stores are present in the Fragment Store and empty if unused. The concrete types, e.g. the position types or read/contig alphabet, can be easily changed by defining a custom config struct which is a template parameter of the Fragment Store class.
Multiple Read Alignment¶
The Fragment Store can represent a multiple read alignment, i.e. is an alignment between the contigs and the set of reads, where one read can be aligned at zero, one or multiple positions of a contig. In the multiple alignment the contig is represented by one line with gaps (-) and the remaining lines are to reads or read segments with gaps aligned to the contig. The following figure shows one contig (blue line at the top) and multiple reads aligned to it arranged as stairs (reads in lower-case align to the reverse strand):
TGAAAACTATATTTATGCTATTCAGTTCTAAATATAGAAATTGAAACAGCTGTGTTTAGTGCCTTTGTTCA-----ACCCCCTTGCAACAACCTTGAGAACCCCAGGGAATTTGTCAATGTCAGGGAAGGAGCATTTTGTCAGTTACCAAATGTGTTTATTACCAG
TGAAAACTATATT ATGCTATTCAGTTCTAAATATAGAAATTGAAACAG GTGTTTAGTGCCTTTGTTCA-----ACCCCCTTGCAACAAC aaccccagggaatttgtcaatgtcagggaaggagc ttttgtcagttaccaaatgtgtttattaccag
tgaa ctatatttatgctattcagttctaaatatagaaatt acagctgtgtttagtgcctttgttca-----acccccttg aacaaccttgagaaccccagggaatttgtcaatgt GGAAGGAGCATTTTGTCAGTTACCAAATGTGTTT TACCAG
TGAAAACTATAT TATGCTATTCAGTTCTAAATATAGAAATTGAAACA ctgtgtttagtgcctttgttca-----acccccttgcaac ACCTTGAGAACCCCAGGGAATTTGTCAATGTCAGG aggagcattttgtcagttaccaaatgtgtttatta at
TGAAAACTATATTTA gctattcagttctaaatatagaaattgaaacagct GTTTAGTGCCTTTGTTCACATAGACCCCCTTGCAA aaccttgagaaccccagggaatttgtcaatgtcag aggagcattttgtcagttaccaaatgtgtttatta AG
TGAAAACTATATTTATGCTATTCAGT GAAATTGAAACAGCTGTGTTTAGTGCCTTTGTTCA ccccttacaacaaccttgagaaccccagggaattt CAGGGAAGGAGCATTTTGTCAGTTACCAAATGTGT G
tgaaaactatatttatgctattcagt GCCTTTGTTCACATAGACCCCCTTGCAACAACCTT cagggaatttgtcaatgtcagggaaggagcatttt CAGTTACCAAATGTGTTTATTACCAG
tgaaaactatatttatgctattcagttcta AG-----ACCCCCTTGCAACAACCTTGAGAACCCCAGGGA ggaaggagcattttgtcagttaccaaatgtgttta
TGAAAACTATATTTATGCTATTCAGTTCTAA A-----ACCCCCTTGCAACAACCTTGAGAACCCCAGGGAA gaaaggagcattttgtcagttaccaaatgtgttta
TGAAAACTATATTTATGCTATTCAGTTCTAAA A-----ACCCCCTTGCAACAACCTTGAGAACCCCAGGGAA AGGAGCATTTTGTCAGTTACCAAATGTGTTTATTA
TGAAAACTATATTTATGCTATTCAGTTCTAAA TGCAACAACCTTGAGAACCCCAGGGAATTTGTCAA ggagcattttgtcagttaccaaatgtgtttattac
TGAAAACTATATTTATGCTATTCAGTTCTAAAT TGCAACAACCTTGAGAACCCCAGGGAATTTGTCAA GGAGCATTTTGTCAGTTACCAAATGTGTTTATTAC
TGAAAACTATATTTATGCTATTCAGTTCTAAAT TGCAACAACCTTGAGAACCCCAGGGAATTTGTCAA GGAGCATTTTGTCAGTTACCAAATGTGTTTATTAT
ctatatttatgctattcagttctaaatatagaaatt tgcaacaaccttgagaaccccagggaatttgtcaa GGAGCATTTTGTCAGTTACCAAATGTGTTTATTAC
ctatatttatgctattcagttctaaatatagaaatt CAACCTTGAGAACCCCAGGGAATTTGTCAATGTCA agcattttgtcagttaccaaatgtgtttattacca
TATTTATGCTATTCAGTTATAAATATAGAAATTGAAACAG CCTTGAGAACCCCAGGGAATTTGTCAATGTCAGGG agcattttgtcagttaccaaatgtgtttattacca
atttatgctattcagttctaaatatagaaattgaa CTTGAGAACCCCAGGGAATTTGTCAATGTCAGGGA GCATTTTGTCAGTTACCAAATGTGTTTATTACCAG
tttacgctattcagtactaaatatagaaattgaaa CTTGAGAACCCCAGGGAATTTGTCAATGTCAGGGA GCATTTTGTCAGTTACCAAATGTGTTTATTACCAG
ttatgctattcagttctaaatatagaaattgaaac gggaatttgtcaatgtcagggaaggagcattttgt AGTTACCAAATGTGTTTATTACCAG
- *Figure 1:* Multiple read alignment
The following figure shows which tables represent the multiple read alignment:
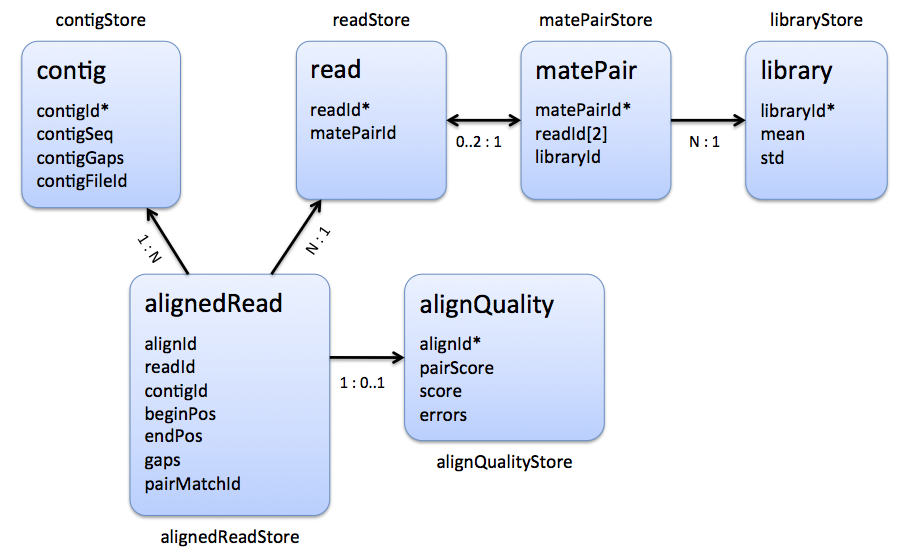
*Figure 2:* Stores used to represent a multiple read alignment
The main table is the alignedReadStore which stores AlignedReadStoreElements. Each entry is an alignment of a read (readId) and a contig (contigId). Introduced gaps are stored as a string of gap anchors in the gaps member of the alignedReadStore entry and the contigStore entry. The begin and end positions of the alignment are given by the beginPos and endPos members which are 0-based positions on the forward strand in gap space, i.e. positions in the gapped contig sequence. If the read is aligned to the reverse strand it holds endPos < beginPos. However, the gaps are always related to the forward strand. Additional information, e.g. the number of errors, an alignment score or additional alignment tags, are stored in the tables alignQualityStore and alignedReadTagStore at position id, where id is a unique id of the AlignedReadStoreElement. Paired-end or mate pair alignments are represented by two entries in the alignedReadStore that have the same pairMatchId value (unequal to INVALID_ID). For orphaned read alignments holds pairMatchId == INVALID_ID.
012345556789 sequence space
012345678901 gap space
contig ACCAC--GTTTG
read1 ACACGGT [2-9[
read2 ACGGTT-G [4-12[
The alignedReadStore is the only store where the id (alignId in the figure) of an element is not implicitly given by its position. The reason for this is that it is necessary in many cases to rearrange the elements of the alignedReadStore, e.g. increasingly by (contigId,beginPos), by readId or pairMatchId. This can be done by sortAlignedReads. If it is necessary to address an element by its id, the elements must be sorted by id first. In the case that ids are not contiguously increasing, e.g. because some elements where removed, they must be renamed by a prior call of compactAlignedReads. Analogously the function compactPairMatchIds renames pairMatchId values contiguously and replaces values that occur in only one alignment by INVALID_ID.
Display Aligned Reads¶
The multiple read alignment can be displayed in text form or in a scalable graphics format (SVG). Therefore first a stairs layout of the reads must be computed via layoutAlignment and stored in an AlignedReadLayout. The function printAlignment can then be used to output a window (beginPos,endPos,firstLine,lastLine) of the read alignment against a contig either to a stream or SVGFile. The following small example demonstrates how to first load two contigs from a Fasta file and then import a read alignment given in SAM format:
#include <iostream>
#include <seqan/store.h>
#include <seqan/misc/misc_svg.h>
using namespace seqan;
int main ()
{
FragmentStore<> store;
loadContigs(store, "ex1.fa");
std::ifstream file("ex1.sam");
read(file, store, Sam());
Then we create a stairs layout of the aligned reads and output a window from gapped position 0 to 150 and line 0 to 36 of the multiple alignments below contig 1 to standard out.
AlignedReadLayout layout;
layoutAlignment(layout, store);
printAlignment(std::cout, Raw(), layout, store, 1, 0, 150, 0, 36);
TTCAAATGAACTTCTGTAATTGAAAAATTCATTTAAGAAATTACAAAATATAGTTGAAAGCTCTAACAATAGACTAAACCAAGCAGAAGAAAGAGGTTCAGAACTTGAAGACAAGTCTCTTATGAATTAACCCAGTCAGACAAAAATAAA
TTCAAATGAACTTCTGTAATTGAAAAATTCATTTAA AATTACAAAATATAGTTGAAAGCTCTAACAATAGA AACCAAGCAGAAGAAAGAGGTTCAGAACTTGAAGA AGTCTCTTATGAATTAACCCAGTCAGACAAAAATA A
TTCAAATGAACTTCTGTAATTGAAAAATTCATTTA GAAATTACAAAATATAGTTGAAAGCTCTAACAATA ACTAAACCAAGCAGAAGAAAGAGGTTCAGAACTTG AGACAAGTCTCTTATGAATTAACCCAGTCAGACAA AAA
ttcaaatgaacttctgtaattgaaaaattcattta GAAATTACAAAATATAGTTGAAAGCTCTAACAATAG AACCAAGCAGAAGAAAGAGGCTCAGAACTTGAAGA AGTCTCTTATGAATTAACCCAGTCAGACAAAAATA A
TCAAATGAACTTCTGTAATTGAAAAATTCATTTAA ATTACAAAATATAGTTGAAAGATCTAACAATAGAC CCAAGCAGAAGAAAGAGGTTCAGAACTTGAAGACAA TTATGAATTAACCCAGTCAGACAAAAATAAA
AATGAACTTCTGTAATTGAAAAATTCATTTAAGAA TTACAAAATATAGTTGAAAGCTCTAACAATAGACT AAGCAGAAGAAAGAGGTTCAGAACTTGAAGACAAG TATGAATTAACCCAGTCAGACAAAAATAAA
ATGAACTTCTGTAATTGAAAAATTCATTTAAGAAA ACAAAATATAGTTGAAAGCTCTAACAATAGACTAA GCAGAAGAAAGAGGTTCAGAACTTGAAGACAAGTC ATGAATTAACCAAGTCAGACAAAAATAAA
ATGAACTTCTGTAATTGAAAAATTCATTTAAGAAA ACAAAATATAGTTGAAAGCTCTAACAATAGACTAA CAGAAGAAAGAGGTTCAGAACTTGAAGACAAGTCT TGAATTAACCCAGTCAGACAAAAATAAA
ATGAACTTCTGTAATTGAAAAATTCATTTAAGAAA ACAAAATATAGTTGAAAGCTCTAACAATAGACTAA CAGAAGAAAGAGGTTCANANNNTGANGACAAGTCT TGAATTAACCCAGTCAGACAAAAATAAA
TGAACTTCTGTAATTGAAAAATTCATTTAAGAAATT CAAAATATAGTTGAAAGCTCTAACAATAGACTAAA GAAGAAAGAGGTTCAGAACTTGAAGACAAGTCTCT GAATTAACCCAGTCAGACAAAAANNAA
TGAACTTCTGTAATTGAAAAATTCATTTAAGAAAT AAAATATAGTTGAAAGCTCTAACAATAGACTAAAC AAGAAAGAGGTTCAGAACTTGAAGACAAGTCTCGT GAATTAACCCAGTCAGACAAAAATAAA
TGAACTTCTGTAATTGAAAAATTCATTTAAGAAAT AAAATATAGTTGAAAGCTCTAACAATAGACTAAAC AAGAAAGAGGTTCAGAACTTGAAGACAAGTCTCTT AATTAACCCAGTCAGACAAAAATAAA
TGAACTTCTGTAATTGAAAAATTCATTTAAGAAAT AAATATAGTTGAAAGCTCTAACAATAGACTAAACC GAAAGAGGTTCAGAACTTGAAGACAAGTCTCTTATG AAA
GAACTTCTGTAATTGAAAAATTCATTTAAGAAATT AAATATAGTTGAAAGCTCTAACAATAGACTAAACC AAGAGGTTCAGAACTTGAAGACAAGTCTCTTATGA
GAACTTCTGTAATTGAAAAATTCATTTAAGAAATT AATATAGTTGAAAGCTCTAACAATAGACTAAACCAA AAGAGGTTCAGAACTTGAAGACAAGTCTCTTATGA
CTTCTGTAATTGAAAAATTCATTTAAGAAATTACA ATATAGTTGAAAGCTCTAACAATAGACTAAACCAA GAGGTTCAGAACTTGAAGACAAGTCTCTTATGAAT
TTCTGTAATTGAAAAATTCATTTAAGAAATTACAA ATAGTTGAAAGCTCTAACAATAGACTAAACCAAGC GAGGTTCAGAACTTGAAGACAAGTCTCTTATGAAT
GTAATTGAAAAATTCATTTAAGAAATTACAAAATA AGTTGAAAGCTCTAACAATAGACTAAACCAAGCAG AGGTTCAGAACTTGAAGACAAGTCTCTTATGAATT
TAATTGAAAAATTCATTTAAGAAATTACAAAATAT TTGAAAGCTCTAACAATAGACTAAACCAAGCAGAA GGTTCAGAACTTGAAGACAAGTCTCTTATGAATTA
AATTGAAAAATTCATTTAAGAAATTACAAAATATA GAAAGCTCTAACAATAGACTAAACCAAGCAGAAGAAAGAG TTCAGAACTTGAAGACAAGTCTCTTATGAATTAAC
AAAATTCATTTAAGAAATTACAAAATATAGTTGAA CTAACAATAGACTAAACCAAGCAGAAGAAAGAGTT CTTGAAGACAAGTCTCTTATGAATTAACCCAGTCA
AAATTCATTTAAGAAATTACAAAATATAGTTGAAA CTAACAATAGACTAAACCAAGCAGAAGAAAGAGGTT TTGAAGACAAGTCTCTTATGAATTAACCCAGTCAG
AATTCATTTAAGAAATTACAAAATATAGTTGAAAG TAACAATAGACTAAACCAAGCAGAAGAAAGAGGTT TGAAGACAAGTCTCTTATGAATTAACCCAGTCAGA
CATTTAAGAAATTACAAAATATAGTTGAAAGCTCT ACAATAGACTAAACCAAGCAGAAGAAAGAGGTTCA TGAAGACAAGTCTCTTATGAATTAACCCAGTCAGACAAAA
TTAAGAAATTACAAAATATAGTTGAAAGCTCTAAC GACTAAACCAAGCAGAAGAAAGAGGTTCAGAACTT AAGACAAGTCTCTTATGAATTAACCCAGTCAGACA
TAAGAAATTACAAAATATAGTTGAAAGCTCTAACAATAGA GGTTCAGAACTTGAAGACAAGTCTCTTATGAATTA
TTACAAAATATAGTTGAAAGCTCTAACAATAGACT GGTTCAGAACTTGAAGACAAGTCTCTTATGAATTA
ATAGTTGAAAGCTCTAACAATAGACTAAACCAAGC GTTCAGAACTTGAAGACAAGTCTCTTATGAATTAAC
AAAGCTCTAACAATAGACTAAACCAAGCAGAAGAA TCAGAACTTGAAGACAAGTCTCTTATGAATTAACC
AAAGCTCTAACAATAGACTAAACCAAGCAGAAGAA NAAGACAAGTCTCTTATGAATTAACCCAGTCAGACA
AAGCTCTAACAATAGACTAAACCAAGCAGAAGAAA GAAGACAAGTCTCTTATGAATTAACCCAGTCAGAC
TAACAATAGACTAAACCAAGCAGAAGAAAGAGGTT AGTCTCTTATGAATTAACCCAGTCAGACAAAAATA
TAACAATAGACTAAACCAAGCAGAAGAAAGAGGTT GTCTCTTATGAATTAACCCAGTCAGACAAAAATAA
AACAATAGACTAAACCAAGCAGAAGAAAGAGGTTC
AACAATAGACTAAACCAAGCAGAAGAAAGAGGTTC
AATAGACTAAACCAAGCAGAAGAAAGAGGTTCAGA
AATAGACTAAACCAAGCAGAAGAAAGAGGTTCAGA
The same window can also be exported as a scalable vector graphic in SVG format (supported by Browsers, Inkscape; see original file]):
{kind=link}
SVGFile svg("layout.svg");
printAlignment(svg, Raw(), layout, store, 1, 0, 150, 0, 36);
return 0;
}

‘’‘Figure 3:’‘’ SVG export of a multiple read alignment
Accessing Pairwise Alignments¶
In the next step, we want to access several pairwise alignments between reads and contig segments. Therefore we first need to get the associated types that the Fragment Store uses to store contig and read sequences and gaps. This can be done by the following typedefs:
typedef Value<TStore::TReadSeqStore>::Type TReadSeq;
typedef Value<TStore::TContigStore>::Type TContig;
typedef Value<TStore::TAlignedReadStore>::Type TAlignedRead;
typedef Gaps<TContig::TContigSeq, AnchorGaps<TContig::TGapAnchors> > TContigGaps;
typedef Gaps<TStore::TReadSeq, AnchorGaps<TAlignedRead::TGapAnchors> > TReadGaps;
TStore::TReadSeq readSeq;
Now we want to extract and output the alignments from the alignedReadStore at position 140,144,...,156. First we store a reference of the alignedRead in ar as we need to access it multiple times. The read sequence is neither stored in the readStore or alignedReadStore as many short sequences can more efficiently be stored in a separate StringSet like the readSeqStore. We copy the read sequence into a local variable (defined outside the loop to save allocations/deallocations) as we need to compute the reverse-complement for reads that align to the reverse strand. Then we create a gaps object that represent the alignment rows of the contig and the aligned read in the multiple sequence alignment. The Gaps object requires references of the sequence and the gap-anchor string stored in the contigStore and the alignedReadStore. We need to limit the view of the contig alignment row to the interval the read aligns to, i.e. the gap position interval [beginPos,endPos[. After that we output both alignment rows.
Tip
The Gaps contains two Holder references to the sequence and the inserted gaps. In our example these Holders are dependent and changes made to the Gaps object like the insertion/deletion of gaps would immediatly be persistent in the Fragment Store.
for (int i = 140; i < 160; i += 4)
{
TAlignedRead &ar = store.alignedReadStore[i];
readSeq = store.readSeqStore[ar.readId];
if (ar.endPos < ar.beginPos)
reverseComplement(readSeq);
TContigGaps contigGaps(
store.contigStore[ar.contigId].seq,
store.contigStore[ar.contigId].gaps);
TReadGaps readGaps(
readSeq,
ar.gaps);
setBeginPosition(contigGaps, std::min(ar.beginPos, ar.endPos));
setEndPosition(contigGaps, std::max(ar.beginPos, ar.endPos));
std::cout << "ALIGNMENT " << i << std::endl;
std::cout << "\tcontig " << ar.contigId << ":\t" << contigGaps;
std::cout << " \t[" << beginPosition(contigGaps) << ".." << endPosition(contigGaps) << "[" << std::endl;
std::cout << "\tread " << ar.readId << ":\t" << readGaps << std::endl;
std::cout << std::endl;
}
ALIGNMENT 140
contig 0: CTGTGTTTAGTGCCTTTGTTCA-----ACCCCCTTGCAAC [266..306[
read 149: CTGTGTTTAGTGCCTTTGTTCA-----ACCCCCTTGCAAC
ALIGNMENT 144
contig 0: AGTGCCTTTGTTCA-----ACCCCCTTGCAACAACC [274..310[
read 153: AGTGCCTTTGTTCACATAGACCCCCTTGCAACAACC
ALIGNMENT 148
contig 0: TTCA-----ACCCCCTTGCAACAACCTTGAGAACCCCAGG [284..324[
read 157: ATAG-----ACCCCCTTGCAACAACCTTGAGAACCCCAGG
ALIGNMENT 152
contig 0: CA-----ACCCCCTTGCAACAACCTTGAGAACCCCAGGGA [286..326[
read 161: CA-----ACCCCCTTGCAACAACCTTGCGAACCCCAGGGA
ALIGNMENT 156
contig 0: CCCCCTTGCAACAACCTTGAGAACCCCAGGGAATT [294..329[
read 165: CCCCCTTGCAACAACCTTGAGAACCCCAGGGAATT
Assignment 1¶
- Type
- Rview
- Objective
- Modify the example above, such that reads that align to the reverse strand are displayed in lower-case letters.
- Difficulty
- Easy
- Hint
- The Dna alphabet used in the fragment store doesn’t support lower-case letters. You have to use a string of chars for readSeq.
- Solution
As we copy the read sequence, it suffices to change the type of the target string readSeq and the sequence type of the read Gaps object into CharString, i.e. a String of char.
typedef Value<TStore::TReadSeqStore>::Type TReadSeq; typedef Value<TStore::TContigStore>::Type TContig; typedef Value<TStore::TAlignedReadStore>::Type TAlignedRead; typedef Gaps<TContig::TContigSeq, AnchorGaps<TContig::TGapAnchors> > TContigGaps; typedef Gaps<CharString, AnchorGaps<TAlignedRead::TGapAnchors> > TReadGaps; CharString readSeq;
Then, we not only need to reverse-complement readSeq if the read aligns to the reverse strand (endPos < beginPos) but also need to convert its letters into lower-case. Therefor SeqAn provides the function toLower. Alternatively, we could iterate over readSeq and add (‘a’-‘A’) to its elements.
for (int i = 140; i < 160; i += 4) { TAlignedRead &ar = store.alignedReadStore[i]; readSeq = store.readSeqStore[ar.readId]; if (ar.endPos < ar.beginPos) { reverseComplement(readSeq); toLower(readSeq); } TContigGaps contigGaps( store.contigStore[ar.contigId].seq, store.contigStore[ar.contigId].gaps); TReadGaps readGaps( readSeq, ar.gaps); setBeginPosition(contigGaps, std::min(ar.beginPos, ar.endPos)); setEndPosition(contigGaps, std::max(ar.beginPos, ar.endPos)); std::cout << "ALIGNMENT " << i << std::endl; std::cout << "\tcontig " << ar.contigId << ":\t" << contigGaps; std::cout << " \t[" << beginPosition(contigGaps) << ".." << endPosition(contigGaps) << "[" << std::endl; std::cout << "\tread " << ar.readId << ":\t" << readGaps << std::endl; std::cout << std::endl; }
Running this program results in the following output.
- ALIGNMENT 140
- contig 0: CTGTGTTTAGTGCCTTTGTTCA—–ACCCCCTTGCAAC [266..306[ read 149: ctgtgtttagtgcctttgttca—–acccccttgcaac
- ALIGNMENT 144
- contig 0: AGTGCCTTTGTTCA—–ACCCCCTTGCAACAACC [274..310[ read 153: AGTGCCTTTGTTCACATAGACCCCCTTGCAACAACC
- ALIGNMENT 148
- contig 0: TTCA—–ACCCCCTTGCAACAACCTTGAGAACCCCAGG [284..324[ read 157: ATAG—–ACCCCCTTGCAACAACCTTGAGAACCCCAGG
- ALIGNMENT 152
- contig 0: CA—–ACCCCCTTGCAACAACCTTGAGAACCCCAGGGA [286..326[ read 161: CA—–ACCCCCTTGCAACAACCTTGCGAACCCCAGGGA
- ALIGNMENT 156
- contig 0: CCCCCTTGCAACAACCTTGAGAACCCCAGGGAATT [294..329[ read 165: cccccttgcaacaaccttgagaaccccagggaatt
Gene Annotation¶
Annotations are represented as a tree that at least contains a root node where all annotations of children or grandchildren of. A typical annotation tree looks as follows:
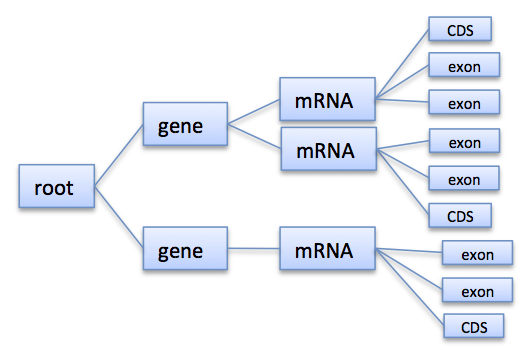
*Figure 4:* Annotation tree example
The following figure shows which tables represent the annotation tree:
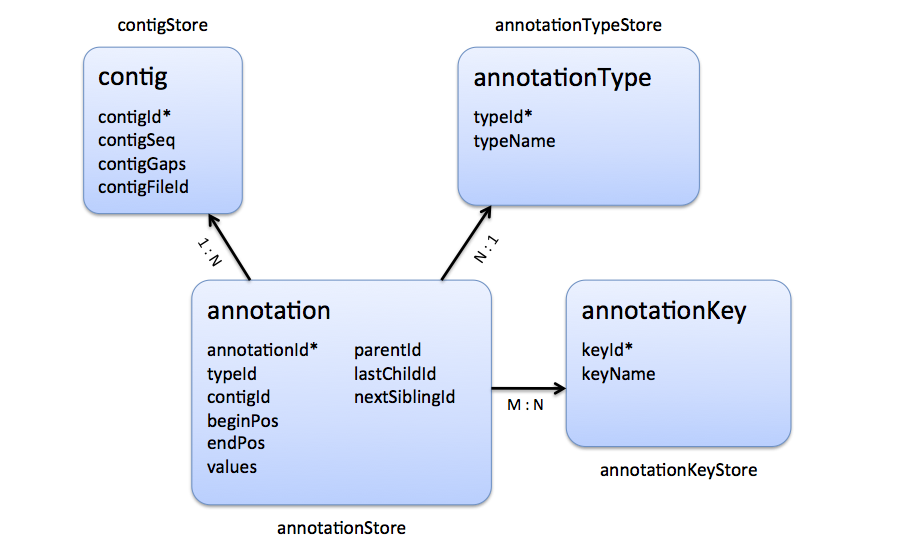
*Figure 5:* Stores involved in gene annotation
Traversing the Annotation Tree¶
The annotation tree can be traversed and accessed with the AnnotationTree Iterator. A new iterator can be created with begin given a reference to the FragmentStore and the tag AnnotationTree:
Iterator<FragmentStore<>, AnnotationTree<> >::Type it;
it = begin(store, AnnotationTree<>());
It starts at the root node and can be moved to adjacent tree nodes with the functions goDown, goUp, and goRight. These functions return a boolean value that indicates whether the iterator could be moved. The functions isLeaf, isRoot, isLastChild return the same boolean without moving the iterator. With goRoot or goTo it can be moved to the root node or an arbitrary node given its annotationId. If the iterator should not be moved but a new iterator at an adjacent nodes is required, the functions nodeDown, nodeUp, nodeRight can be used.
The AnnotationTree iterator supports a preorder DFS traversal and therefore can also be used in typical begin-end loops with the functions goBegin (== goRoot), goEnd, goNext, atBegin, atEnd. During a preorder DFS, the descent into subtree can be skipped by goNextRight, or goNextUp which proceeds with the next sibling or returns to the parent node and proceeds with the next node in preorder DFS.
Accessing the Annotation Tree¶
To access or modify the node an iterator points at, the iterator returns the node’s annotationId by the value function (== operator*). With the annotationId the corresponding entry in the annotationStore could be modified manually or by using convenience functions. The function getAnnotation returns a reference to the corresponding entry in the annotationStore. getName and setName can be used to retrieve or change the identifier of the annotation element. As some annotation file formats don’t give every annotation a name, the function getUniqueName returns the name if non-empty or generates one using the type and id. The name of the parent node in the tree can be determined with getParentName. The name of the annotation type, e.g. ‘mRNA’ or ‘exon’, can be determined and modified with getType and setType.
An annotation can not only reference a region of a contig but also contain additional information given as key-value pairs. The value of a key can be retrieved or set by getValueByKey and assignValueByKeq. The values of a node can be cleared with clearValues.
A new node can be created as first child, last child, or right sibling of the current node with createLeftChile, createRightChild, or createSibling. All three functions return an iterator to the newly created node.
The following tables summarizes the functions provided by the AnnotationTree iterator:
| Function | Description |
|---|---|
| getAnnotation, value | Return annotation object/id of current node |
| [get/set]Name, [get/set]Type | Access name or type of current annotation object |
| clearValues, [get/set]ValueByKey | Access associated values |
| goBegin, goEnd, atBegin, atEnd | Go to or test for begin/end of DFS traversal |
| goNext, goNextRight, goNextUp | go next, skip subtree or siblings during DFS traversal |
| goRoot, goUp, goDown, goRight | Navigate through annotation tree |
| create[Left/Right]Child, createSibling | Create new annotation nodes |
| isRoot, isLeaf | Test for root/leaf node |
File I/O¶
Reads and Contigs¶
To efficiently load reads, use the function loadReads which auto-detects the file format, supporting Fasta, Fastq, QSeq and Raw (see AutoSeqFormat), and uses memory mapping to efficiently load millions of reads, their names and quality values. If not only one but two file names are given, loadReads loads mate pairs or paired-end reads stored in two separate files. Both files are required to contain the same number or reads and reads stored at the same line in both files are interpreted as pairs. The function internally uses appendRead or appendMatePair and reads distributed over multiple files can be loaded with consecutive calls of loadReads.
Contigs can be loaded with the function loadContigs. The function loads all contigs given in a single file or multiple files given a single file name or a StringSet of file names. The function has an additional boolean parameter loadSeqs to load immediately load the contig sequence or if false load the sequence later with loadContig to save memory, given the corresponding contigId. If the contig is accessed by multiple instances/threads the functions lockContig and unlockContig can be used to ensure that the contig is loaded and release it after use. The function unlockAndFreeContig can be used to clear the contig sequence and save memory if the contig is not locked by any instance.
To write all contigs to an open output stream use writeContigs.
Multiple Read Alignments¶
A multiple read alignment can be loaded from an open input stream with:
read(file, store, Sam()); // reads a SAM file
read(file, store, Amos()); // reads a file in the AMOS assembler format
and written to an open output stream with:
write(file, store, Sam()); // writes a SAM file
write(file, store, Amos()); // writes a file in the AMOS assembler format
As SAM supports a multiple read alignment (with padding operations in the CIGAR string) but does not enforce its use. That means that a typical SAM file represents a set of pairwise (not multiple) alignments. To convert all the pairwise alignments into a multiple alignments of all reads, read internally calls the function convertPairWiseToGlobalAlignment. A prior call to loadReads is not necessary (but possible) as SAM contains the read names, sequences and quality values. Contigs can be loaded at any time. If they are not loaded before reading a SAM file, empty sequences are created with the names referred in the SAM file. A subsequent call of loadContigs would load the sequences of these contigs, if they have the same identifier in the contig file.
Annotations¶
A annotation file can be read from an open input stream with:
read(file, store, Gff()); // reads a GFF or GTF file
read(file, store, Ucsc()); // reads a 'knownGene.txt' or 'knownIsoforms.txt' file
The GFF-reader is also able to detect and read GTF files. As the kownGene.txt and knownIsoforms.txt files are two seperate files used by the UCSC Genome Browser, they must be read by two consecutive calls of read (first knownGene.txt then knownIsoforms.txt). An annotation can be loaded without loading the corresponding contigs. In that case empty contigs are created in the contigStore with names given in the annonation. A subsequent call of loadContigs would load the sequences of these contigs, if they have the same identifier in the contig file.
To write an annotation to an open output stream use:
write(file, store, Gff()); // writes a GFF file
write(file, store, Gtf()); // writes a GTF file
Please note, that UCSC files cannot be written due to limitations of the file format.
Stores¶
The Fragment Store consists of the following tables:
Read Stores¶
| Store | Description | Details |
|---|---|---|
| readStore | Reads | String mapping from readId to matePairId |
| readSeqStore | Read sequences | String mapping from readId to readSeq |
| matePairStore | Mate-pairs / pairs of reads | String mapping from matePairId to <readId[2], libId> |
| libraryStore | Mate-pair libraries | String mapping from libId to <mean, std> |
Contig Stores¶
| Store | Description | Details |
|---|---|---|
| contigStore | Contig sequences with gaps | String that maps from contigId to <contigSeq, contigGaps, contigFileId> |
| contigFileStore | Stores information how to load contigs on-demand | String that maps from contigFileId to <fileName, firstContigId> |
Read Alignment Stores¶
| Store | Description | Details |
|---|---|---|
| alignedReadStore | Alignments of reads against contigs | String that stores <alignId, readId, contigId, pairMatchId, beginPos, endPos, gaps> |
| alignedReadTagStore | Additional alignment tags (used in SAM) | String that maps from alignId to alignTag |
| alignQualityStore | Mapping quality of read alignments | String that maps from alignId to <pairScore, score, errors> |
Annotation Stores¶
| Store | Description | Details |
|---|---|---|
| annotationStore | Annotations of contig regions | String that maps from annoId to <contigId, typeId, beginPos, endPos, parentId, lastChildId, nextSiblingId, values> |
Name Stores¶
| annotationNameStore | Annotation names | String that maps from annoId to annoName |
|---|---|---|
| readNameStore | Read identifiers (Fasta ID) | String that maps from readId to readName |
| contigNameStore | Contig identifiers (Fasta ID) | String that maps from contigId to contigName |
| matePairNameStore | Mate-pair identifiers | String that maps from contigId to contigName |
| libraryNameStore | Mate-pair library identifiers | String that maps from libId to libName |