Simple RNA-Seq¶
- Learning Objective
- You will learn how to write a simple gene quantification tool based on RNA-Seq data.
- Difficulty
- Hard
- Duration
- 2h
- Prerequisites
- Genome Annotations, Fragment Store, experience with OpenMP (optional)
RNA-Seq refers to high-throughput sequencing of cDNA in order to get information about the RNA molecules available in a sample. Knowing the sequence and abundance of mRNA allows to determine the (differential) expression of genes, to detect alternative splicing variants, or to annotate yet unknown genes.
In the following tutorial you will develop a simple gene quantification tool. It will load a file containing gene annotations and a file with RNA-Seq read alignments, computes abundances, and outputs RPKM values for each expressed gene.
Albeit its simplicity, this example can be seen as a starting point for more complex applications, e.g. to extend the tool from quantification of genes to the quantification of (alternatively spliced) isoforms, or to de-novo detect yet unannotated isoforms/genes.
You will learn how to use the FragmentStore to access gene annotations and alignments and how to use the IntervalTree to efficiently determine which genes overlap a read alignment.
Introduction to the used Data Structures¶
This section introduces the FragmentStore and the IntervalTree, which are the fundamental data structures used in this tutorial to represent annotations and read alignments and to efficiently find overlaps between them. You may skip one or both subsections if you are already familiar with one or both data structures.
Fragment Store¶
The FragmentStore is a data structure specifically designed for read mapping, genome assembly or gene annotation. These tasks typically require lots of data structures that are related to each other such as
- reads, mate-pairs, reference genome
- pairwise alignments, and
- genome annotation.
The Fragment Store subsumes all these data structures in an easy to use interface. It represents a multiple alignment of millions of reads or mate-pairs against a reference genome consisting of multiple contigs. Additionally, regions of the reference genome can be annotated with features like ‘gene’, ‘mRNA’, ‘exon’, ‘intro’ or custom features. The Fragment Store supports I/O functionality to read/write a read alignment in SAM or AMOS format and to read/write annotations in GFF or GTF format.
The Fragment Store can be compared with a database where each table (called “store”) is implemented as a String member of the FragmentStore class. The rows of each table (implemented as structs) are referred by their ids which are their positions in the string and not stored explicitly. The only exception is the alignedReadStore whose elements of type AlignedReadStoreElement contain an id-member as they may be rearranged in arbitrary order, e.g. by increasing genomic positions or by readId. Many stores have an associated name store to store element names. Each name store is a StringSet that stores the element name at the position of its id. All stores are present in the Fragment Store and empty if unused. The concrete types, e.g. the position types or read/contig alphabet, can be easily changed by defining a custom config struct which is a template parameter of the Fragment Store class.
Annotation Tree¶
Annotations are represented as a tree that at least contains a root node where all annotations are children or grandchildren of. A typical annotation tree looks as follows:
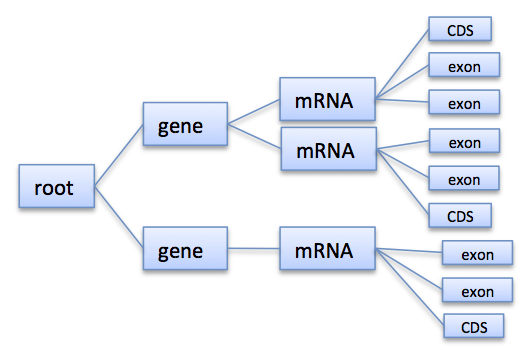
Annotation tree example
In the Fragment Store the tree is represented by annotationStore, annotationTypeStore, annotationKeyStore, and others. Instead of accessing these tables directly, the AnnotationTree Iterator provides a high-level interface to traverse and access the annotation tree.
Interval Tree¶
The IntervalTree is a data structure that stores one-dimensional intervals in a balanced tree and efficiently answers range queries. A range query is an operation that returns all tree intervals that overlap a given query point or interval.
The interval tree implementation provided in SeqAn is based on a Tree which is balanced if all intervals are given at construction time. Interval tree nodes are objects of the IntervalAndCargo class and consist of 2 interval boundaries and additional user-defined information, called cargo. To construct the tree on a set of given interval nodes use the function createIntervalTree. The functions addInterval and removeInterval should only be used if the interval tree needs to be changed dynamically (as they not yet balance the tree).
Import Alignments and Gene Annotations from File¶
At first, our application should create an empty FragmentStore object into which we import a gene annotation file and a file with RNA-Seq alignments. An empty FragmentStore can simply be created with:
FragmentStore<> store;
Files can be read from disk with the function read that expects an open stream (e.g. a STL ifstream), a FragmentStore object, and a File Format tag. The contents of different files can be loaded with subsequent calls of read. As we want the user to specify the files via command line, our application will parse them using the ArgumentParser and store them in an option object.
In your first assignment you need to complete a given code template and implement a function that loads a SAM file and a GTF file into the FragmentStore.
Assignment 1¶
- Type
- Application
- Objective
Use the code template below (click more...) and implement the function loadFiles to load the annotation and alignment files. Use the file paths given in the options object and report an error if the files could not be opened.
#include <iostream> #include <seqan/store.h> #include <seqan/arg_parse.h> #include <seqan/misc/misc_interval_tree.h> #include <seqan/parallel.h> using namespace seqan; // define used types typedef FragmentStore<> TStore; // define options struct Options { std::string annotationFileName; std::string alignmentFileName; }; // // 1. Parse command line and fill Options object // ArgumentParser::ParseResult parseOptions(Options & options, int argc, char const * argv[]) { ArgumentParser parser("gene_quant"); setShortDescription(parser, "A simple gene quantification tool"); setVersion(parser, "1.0"); setDate(parser, "Sep 2012"); addArgument(parser, ArgParseArgument(ArgParseArgument::INPUTFILE)); addArgument(parser, ArgParseArgument(ArgParseArgument::INPUTFILE)); addUsageLine(parser, "[\\fIOPTIONS\\fP] <\\fIANNOTATION FILE\\fP> <\\fIREAD ALIGNMENT FILE\\fP>"); // Parse command line ArgumentParser::ParseResult res = parse(parser, argc, argv); if (res == ArgumentParser::PARSE_OK) { // Extract option values getArgumentValue(options.annotationFileName, parser, 0); getArgumentValue(options.alignmentFileName, parser, 1); } return res; } // // 2. Load annotations and alignments from files // bool loadFiles(TStore & store, Options const & options) { // INSERT YOUR CODE HERE ... // return true; } int main(int argc, char const * argv[]) { Options options; TStore store; ArgumentParser::ParseResult res = parseOptions(options, argc, argv); if (res != ArgumentParser::PARSE_OK) return res == ArgumentParser::PARSE_ERROR; if (!loadFiles(store, options)) return 1; return 0; }
- Hint
- Open STL std::fstream objects and use the function read with a SAM or GTF tag.
- ifstream::open requires the file path to be given as a C-style string (const char *).
- Use string::c_str to convert the option strings into C-style strings.
- The function read expects a stream, a FragmentStore and a tag, i.e. Sam() or Gtf().
- Solution
// // 2. Load annotations and alignments from files // bool loadFiles(TStore & store, Options const & options) { std::ifstream alignmentFile(options.alignmentFileName.c_str()); if (!alignmentFile.good()) { std::cerr << "Couldn't open alignment file " << options.alignmentFileName << std::endl; return false; } std::cerr << "Loading read alignments ..... " << std::flush; read(alignmentFile, store, Sam()); std::cerr << "[" << length(store.alignedReadStore) << "]" << std::endl; // load annotations std::ifstream annotationFile(options.annotationFileName.c_str()); if (!annotationFile.good()) { std::cerr << "Couldn't open annotation file" << options.annotationFileName << std::endl; return false; } std::cerr << "Loading genome annotation ... " << std::flush; read(annotationFile, store, Gtf()); std::cerr << "[" << length(store.annotationStore) << "]" << std::endl; return true; }
Extract Gene Intervals¶
Now that the Fragment Store contains the whole annotation tree, we want to traverse the genes and extract the genomic ranges they span. In the annotation tree, genes are (the only) children of the root node. To efficiently retrieve the genes that overlap read alignments later, we want to use interval trees, one for each contig. To construct an interval tree, we first need to collect IntervalAndCargo objects in a string and pass them to createIntervalTree. See the interval tree demo in core/demos/interval_tree.cpp for more details. As cargo we use the gene’s annotation id to later retrieve all gene specific information. The strings of IntervalAndCargo objects should be grouped by contigId and stored in an (outer) string of strings. For the sake of simplicity we don’t differ between genes on the forward or reverse strand and instead always consider the corresponding intervals on the forward strand.
To define this string of strings of IntervalAndCargo objects, we first need to determine the types used to represent an annotation. All annotations are stored in the annotationStore which is a Fragment Store member and whose type is TAnnotationStore. The value type of the annotation store is the class AnnotationStoreElement. Its member typedefs TPos and TId define the types it uses to represent a genomic position or the annotation or contig id:
typedef FragmentStore<> TStore;
typedef Value<TStore::TAnnotationStore>::Type TAnnotation;
typedef TAnnotation::TId TId;
typedef TAnnotation::TId TPos;
typedef IntervalAndCargo<TPos, TId> TInterval;
The string of strings of intervals can now be defined as:
String<String<TInterval> > intervals;
In your second assignment you should use an AnnotationTree Iterator annotation tree iterator] to traverse all genes in the annotation tree. For each gene, determine its genomic range (projected to the forward strand) and add a new TInterval object to the intervals[contigId] string, where contigId is the id of the contig containing that gene.
Assignment 2¶
- Type
- Application
- Objective
Use the code template below (click more..). Implement the function extractGeneIntervals that should extract genes from the annotation tree (see AnnotationTree Iterator) and create strings of IntervalAndCargo objects - one for each config - that contains the interval on the forward contig strand and the gene’s annotation id.
Extend the definitions:
// define used types typedef FragmentStore<> TStore; typedef Value<TStore::TAnnotationStore>::Type TAnnotation; typedef TAnnotation::TId TId; typedef TAnnotation::TPos TPos; typedef IntervalAndCargo<TPos, TId> TInterval;
Add a function:
// // 3. Extract intervals from gene annotations (grouped by contigId) // void extractGeneIntervals(String<String<TInterval> > & intervals, TStore const & store) { // INSERT YOUR CODE HERE ... // }
Extend the main function:
TStore store; String<String<TInterval> > intervals;
and
if (!loadFiles(store, options)) return 1; extractGeneIntervals(intervals, store);
- Hint
- You can assume that all genes are children of the root node, i.e. create an AnnotationTree Iterator, [go down to the first gene and go right to visit all other genes. Use getAnnotation to access the gene annotation and value to get the annotation id.
Make sure that you append IntervalAndCargo objects, where i1 < i2 holds, as opposed to annotations where beginPos > endPos is possible. Remember to ensure that intervals is of appropriate size, e.g. with
resize(intervals, length(store.contigStore));
Use appendValue to add a new TInverval object to the inner string, see IntervalAndCargo constructor for the constructor.
- Solution
// // 3. Extract intervals from gene annotations (grouped by contigId) // void extractGeneIntervals(String<String<TInterval> > & intervals, TStore const & store) { // extract intervals from gene annotations (grouped by contigId) resize(intervals, length(store.contigStore)); Iterator<TStore const, AnnotationTree<> >::Type it = begin(store, AnnotationTree<>()); if (!goDown(it)) return; do { SEQAN_ASSERT_EQ(getType(it), "gene"); TPos beginPos = getAnnotation(it).beginPos; TPos endPos = getAnnotation(it).endPos; TId contigId = getAnnotation(it).contigId; if (beginPos > endPos) std::swap(beginPos, endPos); // insert forward-strand interval of the gene and its annotation id appendValue(intervals[contigId], TInterval(beginPos, endPos, value(it))); } while (goRight(it)); }
Construct Interval Trees¶
With the strings of gene intervals - one for each contig - we now can construct interval trees. Therefore, we specialize an IntervalTree with the same position and cargo types as used for the IntervalAndCargo objects. As we need an interval tree for each contig, we instantiate a string of interval trees:
typedef IntervalTree<TPos, TId> TIntervalTree;
String<TIntervalTree> intervalTrees;
Your third assignment is to implement a function that constructs the interval trees for all contigs given the string of interval strings.
Assignment 3¶
- Type
- Application
- Objective
Use the code template below (click more...). Implement the function constructIntervalTrees that uses the interval strings to construct for each contig an interval tree. Optional: Use OpenMP to parallelize the construction over the contigs, see SEQAN_OMP_PRAGMA.
Extend the definitions:
// define used types typedef FragmentStore<> TStore; typedef Value<TStore::TAnnotationStore>::Type TAnnotation; typedef TAnnotation::TId TId; typedef TAnnotation::TPos TPos; typedef IntervalAndCargo<TPos, TId> TInterval; typedef IntervalTree<TPos, TId> TIntervalTree;
Add a function:
// // 4. Construct interval trees // void constructIntervalTrees(String<TIntervalTree> & intervalTrees, String<String<TInterval> > & intervals) { // INSERT YOUR CODE HERE ... // }
Extend the main function:
String<String<TInterval> > intervals; String<TIntervalTree> intervalTrees;
and
extractGeneIntervals(intervals, store); constructIntervalTrees(intervalTrees, intervals);
- Hint
First, resize the string of interval trees accordingly:
resize(intervalTrees, length(intervals));
- Hint
Use the function createIntervalTree.
Optional: Construct the trees in parallel over all contigs with an OpenMP parallel for-loop, see here for more information about OpenMP.
- Solution
// // 4. Construct interval trees // void constructIntervalTrees(String<TIntervalTree> & intervalTrees, String<String<TInterval> > & intervals) { int numContigs = length(intervals); resize(intervalTrees, numContigs); SEQAN_OMP_PRAGMA(parallel for) for (int i = 0; i < numContigs; ++i) createIntervalTree(intervalTrees[i], intervals[i]); }
Compute Gene Coverage¶
To determine gene expression levels, we first need to compute the read coverage, i.e. the total number of reads overlapping a gene. Therefore we use a string of counters addressed by the annotation id.
String<unsigned> readsPerGene;
For each read alignment we want to determine the overlapping genes by conducting a range query via findIntervals and then increment their counters by 1. To address the counter of a gene, we use its annotation id stored as cargo in the interval tree.
Read alignments are stored in the alignedReadStore, a string of AlignedReadStoreElements objects. Their actual type can simply be determined as follows:
typedef Value<TStore::TAlignedReadStore>::Type TAlignedRead;
Given the contigId, beginPos, and endPos we will retrieve the annotation ids of overlapping genes from the corresponding interval tree.
Your fourth assignment is to implement the count function that performs all the above described steps. Optionally, use OpenMP to parallelize the counting.
Assignment 4¶
- Type
- Application
- Objective
Use the code template below (click more...). Implement the function countReadsPerGene that counts for each gene the number of overlapping reads. Therefore determine for each AlignedReadStoreElement begin and end positions (on forward strand) of the alignment and increment the readsPerGene counter for each overlapping gene.
Optional: Use OpenMP to parallelize the function, see SEQAN_OMP_PRAGMA.
Extend the definitions:
// define used types typedef FragmentStore<> TStore; typedef Value<TStore::TAnnotationStore>::Type TAnnotation; typedef TAnnotation::TId TId; typedef TAnnotation::TPos TPos; typedef IntervalAndCargo<TPos, TId> TInterval; typedef IntervalTree<TPos, TId> TIntervalTree; typedef Value<TStore::TAlignedReadStore>::Type TAlignedRead;
Add a function:
// // 5. Count reads per gene // void countReadsPerGene(String<unsigned> & readsPerGene, String<TIntervalTree> const & intervalTrees, TStore const & store) { // INSERT YOUR CODE HERE ... // }
Extend the main function:
String<TIntervalTree> intervalTrees; String<unsigned> readsPerGene;
and
extractGeneIntervals(intervals, store); constructIntervalTrees(intervalTrees, intervals); countReadsPerGene(readsPerGene, intervalTrees, store);
- Hint
resize(readsPerGene, length(store.annotationStore), 0);
Make sure that you search with findIntervals where query_begin < query_end holds, as opposed to read alignments where beginPos > endPos is possible.
- Hint
The result of a range query is a string of annotation ids given to findIntervals by-reference:
String<TId> result;
Reuse the result string for multiple queries (of the same thread, use private(result) for OpenMP).
- Solution
// // 5. Count reads per gene // void countReadsPerGene(String<unsigned> & readsPerGene, String<TIntervalTree> const & intervalTrees, TStore const & store) { resize(readsPerGene, length(store.annotationStore), 0); String<TId> result; int numAlignments = length(store.alignedReadStore); // iterate aligned reads and get search their begin and end positions SEQAN_OMP_PRAGMA(parallel for private(result)) for (int i = 0; i < numAlignments; ++i) { TAlignedRead const & ar = store.alignedReadStore[i]; TPos queryBegin = _min(ar.beginPos, ar.endPos); TPos queryEnd = _max(ar.beginPos, ar.endPos); // search read-overlapping genes findIntervals(intervalTrees[ar.contigId], queryBegin, queryEnd, result); // increase read counter for each overlapping annotation given the id in the interval tree for (unsigned j = 0; j < length(result); ++j) { SEQAN_OMP_PRAGMA(atomic) readsPerGene[result[j]] += 1; } } }
Output RPKM Values¶
In the final step, we want to output the gene expression levels in a normalized measure. We therefore use RPKM values, i.e. the number of reads per kilobase of exon model per million mapped reads (1). One advantage of RPKM values is their independence of the sequencing throughput (normalized by total mapped reads), and that they allow to compare the expression of short with long transcripts (normalized by exon length).
The exon length of an mRNA is the sum of lengths of all its exons. As a gene may have multiple mRNA, we will simply use the maximum of all their exon lengths.
Your final assignment is to output the RPKM value for genes with a read counter > 0. To compute the exon length of the gene (maximal exon length of all mRNA) use an AnnotationTree Iterator and iterate over all mRNA (children of the gene) and all exons (children of mRNA). For the number of total mapped reads simply use the number of alignments in the alignedReadStore. Output the gene names and their RPKM values separated by tabs as follows:
#gene name RPKM value
ENSMUSG00000053211 5932.12
ENSMUSG00000069053 10540.1
ENSMUSG00000056673 12271.3
ENSMUSG00000069049 10742.2
ENSMUSG00000091749 7287.66
ENSMUSG00000068457 37162.8
ENSMUSG00000069045 13675
ENSMUSG00000069044 6380.36
ENSMUSG00000077793 2088.62
ENSMUSG00000000103 7704.74
ENSMUSG00000091571 10965.2
ENSMUSG00000069036 127128
ENSMUSG00000090405 10965.2
ENSMUSG00000090652 35271.2
ENSMUSG00000052831 68211.2
ENSMUSG00000069031 37564.2
ENSMUSG00000071960 34984
ENSMUSG00000091987 37056.3
ENSMUSG00000090600 2310.18
Download and decompress the attached mouse annotation ([raw-attachment:Mus_musculus.NCBIM37.61.gtf.zip Mus_musculus.NCBIM37.61.gtf.zip]) and the alignment file of RNA-Seq reads aligned to chromosome Y ([raw-attachment:sim40mio_onlyY.sam.zip sim40mio_onlyY.sam.zip]). Test your program and compare your output with the output above.
Assignment 5¶
- Type
- Application
- Objective
Use the code template below (click more...). Implement the function outputGeneCoverage that outputs for each expressed gene the gene name and the expression level as RPKM as tab-separated values.
Add a function:
// // 6. Output RPKM values // void outputGeneCoverage(String<unsigned> const & readsPerGene, TStore const & store) { // INSERT YOUR CODE HERE ... // }
Extend the main function:
extractGeneIntervals(intervals, store); constructIntervalTrees(intervalTrees, intervals); countReadsPerGene(readsPerGene, intervalTrees, store); outputGeneCoverage(readsPerGene, store);
- Hint
- To compute the maximal exon length use three nested loops: (1) enumerate all genes, (2) enumerate all mRNA of the gene, and (3) enumerate all exons of the mRNA and sum up their lengths.
- Hint
- Remember that exons are not the only children of mRNA.
- Solution
// // 6. Output RPKM values // void outputGeneCoverage(String<unsigned> const & readsPerGene, TStore const & store) { // output abundances for covered genes Iterator<TStore const, AnnotationTree<> >::Type transIt = begin(store, AnnotationTree<>()); Iterator<TStore const, AnnotationTree<> >::Type exonIt; double millionMappedReads = length(store.alignedReadStore) / 1000000.0; std::cout << "#gene name\tRPKM value" << std::endl; for (unsigned j = 0; j < length(readsPerGene); ++j) { if (readsPerGene[j] == 0) continue; unsigned mRNALengthMax = 0; goTo(transIt, j); // determine maximal mRNA length (which we use as gene length) SEQAN_ASSERT_NOT(isLeaf(transIt)); goDown(transIt); do { exonIt = nodeDown(transIt); unsigned mRNALength = 0; // determine mRNA length, sum up the lengths of its exons do { if (getAnnotation(exonIt).typeId == store.ANNO_EXON) mRNALength += abs((int)getAnnotation(exonIt).beginPos - (int)getAnnotation(exonIt).endPos); } while (goRight(exonIt)); if (mRNALengthMax < mRNALength) mRNALengthMax = mRNALength; } while (goRight(transIt)); // RPKM is number of reads mapped to a gene divided by its gene length in kbps // and divided by millions of total mapped reads std::cout << store.annotationNameStore[j] << '\t'; std::cout << readsPerGene[j] / (mRNALengthMax / 1000.0) / millionMappedReads << std::endl; } }
Next Steps¶
- See [MWM+08] for further reading.
- Read the Basic SAM and BAM I/O Tutorial and change your program to stream a SAM file instead of loading it as a whole.
- Change the program such that it attaches the RPKM value as a key-value pair (see assignValueByKey) to the annotation of each gene and output a GFF file.
- Continue with the Tutorial rest of the tutorials]].