Alignment Representation¶
- Learning Objective
- This tutorial introduces you to the two data structures that can be used to represent an alignment in SeqAn. You will learn basic techniques to create and modify such data structures and how to access certain information from these data structures.
- Difficulty
- Basic
- Duration
- 0:45h
- Prerequisites
- A First Example, Alphabets, Sequences, String Sets, Iterators
Before we want to explain SeqAn’s alignment algorithms in detail, we will give you an insight in the underlying data structures that are used to actually represent an alignment in SeqAn. First, we put our focus on the possible representations of alignments and the ways to access and edit different information of an alignment. The two main objects for this purpose are the Align and the Alignment Graph data structure.
Align Data Structure¶
The Align data structure is simply a set of multiple Gaps data structures. A Gaps data structure is a container storing gap information for a given source sequence. The gap information is put on top of the source sequence (coordinates of the gapped sequence refer to the gap space) without directly applying them to the source (coordinates of the ungapped sequence refer to the source space). This way operating with gaps sustains very flexible.
There are two specializations available for the Gaps data structures: Array Gaps and Anchor Gaps. They differ in the way they implement the gap space.
Note
In general, using Array Gaps is sufficient for most applications. This specialization is also the default one if nothing else is specified. It simply uses an array which stores the counts of gaps and characters in an alternating order. Thus, it is quite efficient to extend existing gaps while it is more expensive to search within the gapped sequence or insert new gaps. Alternatively, one should prefer Anchor Gaps if many conversions between coordinates of the gap and the source space are needed as binary search can be conducted to search for specific positions.
Now, let’s start by constructing our first alignment. Before we can make use of any of the mentioned data structures, we need to tell the program where to find the definitions. This can be achieved by including the header file <seqan/align.h> which contains the necessary data structures and functions associated with the alignments. The next steps would be to implement the main function of our program and to define the types that we want to use.
#include <iostream>
#include <seqan/align.h>
using namespace seqan;
int main()
{
We first define the type of the input sequences (TSequence). Then we can define the type of our actual Align object we want to use. In an Align object, the gapped sequences are arranged in rows. You can use the Metafunction Row to get the correct type of the used Gaps objects. In the following we use the term row to explicitly refer to a gapped sequence as a member of the Align object. We will use the term gapped sequence to describe functionalities that is related to the Gaps data structure independent of the Align object.
typedef String<char> TSequence; // sequence type
typedef Align<TSequence,ArrayGaps> TAlign; // align type
typedef Row<TAlign>::Type TRow; // gapped sequence type
After defining the types, we can continue to actually construct our own Align object. Therefore, we need to resize the alignment object in order to reserve space for the sequences we want to add. In our case, we assume a pairwise alignment, hence we reserve space for 2 sequences. With the function row, we get access to the gapped sequence at a specific row in the alignment object. This is similar to the value function used in String Sets. Now, we can assign the source to the corresponding gapped sequence.
TSequence seq1 = "CDFGDC";
TSequence seq2 = "CDEFGAHGC";
TAlign align;
resize(rows(align), 2);
assignSource(row(align,0),seq1);
assignSource(row(align,1),seq2);
After assigning the sources to the gapped sequences, we need to add some gaps to make it to look like a real alignment. You can use the functions insertGap() and removeGap to insert and delete one gap or insertGaps() and removeGaps to insert and delete multiple gaps in a gapped sequence.
std::cout << align;
TRow &row1 = row(align,0);
TRow &row2 = row(align,1);
insertGap(row1,2);
insertGaps(row1,5,2);
std::cout << align;
Congratulations! You have created your first alignment. Note that we used a reference declaration TRow & for the variables row1 and row2. Without the reference, we would only modify copies of rows and the changes would not effect our align object.
Gap Space vs. Source Space¶
In the next steps, we want to dig a little deeper to get a feeling for the gap space and the source space. As mentioned above, the gaps are not inserted into the source but put on top of them in a separate space, the gap space. When inserting gaps, the gap space is modified and all coordinates right of the inserted gap are shifted to the right by the size of the gap. At the same time, the coordinates of the source remain unchanged. Using the function toSourcePosition(), we can determine to which position in the source space our current position in the gapped sequence (gap space) maps.
std::cout << std::endl << "ViewToSource1: ";
for(unsigned i = 0; i < length(row1); ++i)
std::cout << toSourcePosition(row1, i) << ",";
std::cout << std::endl << "ViewToSource2: ";
for(unsigned i = 0; i < length(row2); ++i)
std::cout << toSourcePosition(row2, i) << ",";
std::cout << std::endl;
If the position in the gap space is actually a gap, then toSourcePosition() returns the source position of the next character to the right that is not a gap. Vice versa, we can determine where our current source position maps into the gap space using the function toViewPosition().
std::cout << std::endl << "SourceToView1: ";
for(unsigned i = 0; i < length(source(row1)); ++i)
std::cout << toViewPosition(row1, i) << ",";
std::cout << std::endl << "SourceToView2: ";
for(unsigned i = 0; i < length(source(row2)); ++i)
std::cout << toViewPosition(row2, i) << ",";
std::cout << std::endl;
And here is the output of this short example program so far:
0 .
CDFGDC
||
CDEFGA
0 .
CD-FG--DC
|| || |
CDEFGAHGC
ViewToSource1: 0,1,2,2,3,4,4,4,5,
ViewToSource2: 0,1,2,3,4,5,6,7,8,
SourceToView1: 0,1,3,4,7,8,
SourceToView2: 0,1,2,3,4,5,6,7,8,
In the first alignment, it seems that the end of the second row is cropped off to match the size of the first one. This effect takes place only in the visualization but is not explicitly applied to the gapped sequence. The second alignment is the one we manually constructed. Here, you can see that the second row is expanded to its full size while it matches the size of the first row. However, it is possible to explicitly crop off the ends of a gapped sequence by using the functions setClippedBeginPosition and setClippedEndPosition. These functions shrink the gap space and can be understood as defining an infix of the gapped sequence. After the clipping, the relative view position changes according to the clipping and so does the mapping of the source positions to the gap space. The mapping of the view positions to the source space does not change.
std::cout << std::endl << "Before clipping:\n" << align;
setClippedBeginPosition(row1,1);
setClippedEndPosition(row1,7);
setClippedBeginPosition(row2,1);
setClippedEndPosition(row2,7);
std::cout << std::endl << "After clipping:\n" << align;
std::cout << std::endl << "ViewToSource1: ";
for(unsigned i = 0; i < length(row1); ++i)
std::cout << toSourcePosition(row1, i) << ",";
std::cout << std::endl << "ViewToSource2: ";
for(unsigned i = 0; i < length(row2); ++i)
std::cout << toSourcePosition(row2, i) << ",";
std::cout << std::endl;
std::cout << std::endl << "SourceToView1: ";
for(unsigned i = 0; i < length(source(row1)); ++i)
std::cout << toViewPosition(row1, i) << ",";
std::cout << std::endl << "SourceToView2: ";
for(unsigned i = 0; i < length(source(row2)); ++i)
std::cout << toViewPosition(row2, i) << ",";
std::cout << std::endl;
Here the output of the clipping procedure.
Before clipping:
0 .
CD-FG--DC
|| || |
CDEFGAHGC
After clipping:
0 .
D-FG--
| ||
DEFGAH
ViewToSource1: 1,2,2,3,4,4,
ViewToSource2: 1,2,3,4,5,6,
SourceToView1: -1,0,2,3,6,7,
SourceToView2: -1,0,1,2,3,4,5,6,7,
Note
It is important to understand the nature of the clipping information. It virtually shrinks the gap space not physically. That means the information before/after the begin/end of the clipping still exists and the physical gap space remains unchanged. To the outer world it seems the alignment is cropped off irreparably. But you can expand the alignment again by resetting the clipping information.
Iterating over Gapped Sequences¶
In the last part of this section, we are going to iterate over a Gaps object. This can be quite useful if one needs to parse the alignment rows to access position specific information. First, we have to define the type of the Iterator, which can be easily done by using the metafunction Iterator. Remember that we iterate over an TRow object. Then we have to construct the iterators it which points to the begin of row1 using the begin function and itEnd which points behind the last value of row1 using the end function. If you need to refresh the Iterator Concept you can read the Tutorial Iterators. While we iterate over the gapped sequence, we can ask if the current value, at which the iterator it points to, is a gap or not by using the function isGap(). Use gapValue to print the correct gap symbol.
typedef Iterator<TRow>::Type TRowIterator;
TRowIterator it = begin(row1);
TRowIterator itEnd = end(row1);
for(; it != itEnd; ++it)
{
if(isGap(it))
std::cout << gapValue<char>();
else
std::cout << *it;
}
std::cout << std::endl;
We will now reset the clipping of row1 using clearClipping and iterate again over it to see its effect.
clearClipping(row1);
it = begin(row1);
itEnd = end(row1);
for(; it != itEnd; ++it)
{
if(isGap(it))
std::cout << gapValue<char>();
else
std::cout << *it;
}
std::cout << std::endl;
return 0;
}
D-FG--
CD-FG--DC
Here you can see how resetting the clipping positions brings back our complete row.
Assignment 1¶
- Type
- Review
- Objective
- Construct an alignment using the Align data structure for the sequences "ACGTCACCTC" and "ACGGGCCTATC". Insert two gaps at the second position and insert one gap at the fifth position of the first sequence. Insert one gap at the ninth position of the second sequence. Iterate over the rows of your Align object and print the total count of gaps that exist within the alignment.
- Hints
- You can use the function countGaps to count the number of consecutive gaps starting from the current position of the iterator.
- Solution
#include <iostream> #include <seqan/align.h> using namespace seqan; int main() { // Defining all types that are needed. typedef String<char> TSequence; typedef Align<TSequence,ArrayGaps> TAlign; typedef Row<TAlign>::Type TRow; typedef Iterator<TRow>::Type TRowIterator; TSequence seq1 = "ACGTCACCTC"; TSequence seq2 = "ACGGGCCTATC"; // Initializing the align object. TAlign align; resize(rows(align), 2); assignSource(row(align,0),seq1); assignSource(row(align,1),seq2); // Use references to the rows of align. TRow & row1 = row(align,0); TRow & row2 = row(align,1); // Insert gaps. insertGaps(row1,2,2); insertGap(row1,7); // We need to pass the view position which is changed due to the previous insertion. insertGaps(row2,9,2); // Initialize the row iterators. TRowIterator itRow1 = begin(row1); TRowIterator itEndRow1 = end(row1); TRowIterator itRow2 = begin(row2); // Iterate over both rows simultaneously. int gapCount = 0; for(;itRow1 != itEndRow1; ++itRow1, ++itRow2) { if(isGap(itRow1)) { gapCount += countGaps(itRow1); // Count the number of consecutive gaps from the current position in row1. itRow1 += countGaps(itRow1); // Jump to next position to check for gaps. itRow2 += countGaps(itRow1); // Jump to next position to check for gaps. } if(isGap(itRow2)) { gapCount += countGaps(itRow2); // Count the number of consecutive gaps from the current position in row2. itRow1 += countGaps(itRow2); // Jump to next position to check for gaps. itRow2 += countGaps(itRow2); // Jump to next position to check for gaps. } } // Print the result. std::cout << "Number of gaps: " << gapCount << std::endl; }
AlignmentGraph Data Structure¶
Another very useful representation of alignments is given by the Alignment Graph. It is a graph in which each vertex corresponds to a sequence segment, and each edge indicates an ungapped alignment between the connected vertices, or more precisely between the sequences stored in those vertices. Here is an example of such a graph:
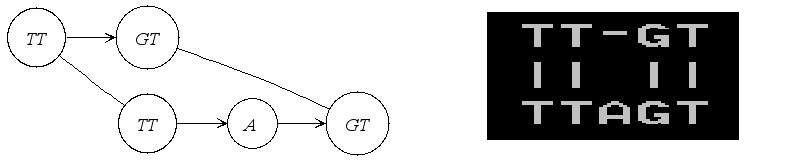
In the following we will actually construct this example step by step. First we include the iostream header from the STL and the <seqan/align.h> header to include all necessary functions and data structures we want to use. We use the namespace seqan and write the main function with an empty body.
#include <iostream>
#include <seqan/align.h>
using namespace seqan;
int main()
{
At the begin of the function we define our types we want to use later on. We define TSequence as the type of our input strings. Since we work with a Dna alphabet we define TSequence as a String over a Dna alphabet. For the AlignmentGraph we need two StringSets. The TStringSet is used to actually store the input sequences and the TDepStringSet is internally used by the AlignmentGraph. That is the AlignmentGraph does not copy the sources into its data structure but rather stores a reference to each of the given input strings as it does not modify the input sequences. The Dependent StringSet facilitates this behavior. In the end we define the actual AlignmentGraph type.
typedef String<Dna> TSequence;
typedef StringSet<TSequence> TStringSet;
typedef StringSet<TSequence, Dependent<> > TDepStringSet;
typedef Graph<Alignment<TDepStringSet> > TAlignGraph;
We first create our two input sequences TTGT and TTAGT append them to the StringSet strings using the appendValue function and pass the initialized strings object as a parameter to the constructor of the AlignmentGraph alignG.
TSequence seq1 = "TTGT";
TSequence seq2 = "TTAGT";
TStringSet strings;
appendValue(strings, seq1);
appendValue(strings, seq2);
TAlignGraph alignG(strings);
Before we construct the alignment we print the unmodified AlignmentGraph. Then we add some alignment information to the graph. In order to add an ungapped alignment segment we have to add an edge connecting two nodes of different input sequences. To do so we can use the function addEdge and specify the two vertices that should be connected. Since we do not have any vertices yet, we create them on the fly using the function addVertex. The function addVertex gets as second parameter the id which points to the the correct input sequence within the strings object. We can use the function positionToId() to receive the id that corresponds to a certain position within the underlying Dependent StringSet of the AlignmentGraph. We can access the Dependent StringSet using the function stringSet(). The third parameter of addVertex specifies the begin position of the segment within the respective input sequence and the fourth parameter specifies its length. Now, we add an edge between the two vertices of each input sequence which covers the first two positions. In the next step we have to add a gap. We can do this simply by just adding a vertex that covers the inserted string. Finally we have to add the second edge to represent the last ungapped sequence and then we print the constructed alignment.
::std::cout << alignG << ::std::endl;
addEdge(alignG, addVertex(alignG, positionToId(stringSet(alignG),0), 0, 2),
addVertex(alignG, positionToId(stringSet(alignG),1), 0, 2));
addVertex(alignG, positionToId(stringSet(alignG), 1),2,1);
addEdge(alignG, addVertex(alignG, positionToId(stringSet(alignG),0), 2, 2),
addVertex(alignG, positionToId(stringSet(alignG),1), 3, 2));
::std::cout << alignG << ::std::endl;
return 0;
}
Here the output of the program. The first output prints the empty adjacency and edge list. The second output prints our desired alignment.
Adjacency list:
Edge list:
Alignment matrix:
0 .
TT-GT
|| ||
TTAGT
The general usage of graphs is explained in the Graphs tutorial.
Assignment 2¶
- Type
- Review
- Objective
- Construct a multiple sequence alignment using the Alignment Graph data structure. Use the three sequences GARFIELDTHECAT, GARFIELDTHEBIGCAT and THEBIGCAT and align them such that you obtain the maximal number of matches.
- Hints
- The function findVertex returns the vertex of an AlignmentGraph that covers the given position in the given sequence.
- Solution
#include <iostream> #include <seqan/align.h> using namespace seqan; int main() { // Define the types we need. typedef String<char> TSequence; typedef StringSet<TSequence> TStringSet; typedef StringSet<TSequence, Dependent<> > TDepStringSet; typedef Graph<Alignment<TDepStringSet> > TAlignGraph; // Initializing the sequences and the string set. TSequence seq1 = "GARFIELDTHECAT"; TSequence seq2 = "GARFIELDTHEBIGCAT"; TSequence seq3 = "THEBIGCAT"; TStringSet strings; appendValue(strings, seq1); appendValue(strings, seq2); appendValue(strings, seq3); // Load the string set into the Alignment Graph. TAlignGraph alignG(strings); // Add two vertices covering "GARFIELD" in the first and the second sequence and connect them with an edge. addEdge(alignG, addVertex(alignG, positionToId(stringSet(alignG),0), 0, 8), addVertex(alignG, positionToId(stringSet(alignG),1), 0, 8)); // Add two vertices covering "THE" in the first and the second sequence and connect them with an edge. addEdge(alignG, addVertex(alignG, positionToId(stringSet(alignG),0), 8, 3), addVertex(alignG, positionToId(stringSet(alignG),1), 8, 3)); // Find the vertex covering "THE" in the first sequence and add the vertex covering "THE" in the third sequence and connect them with an edge. addEdge(alignG, findVertex(alignG, positionToId(stringSet(alignG),0), 8), addVertex(alignG, positionToId(stringSet(alignG),2), 0, 3)); // Find the vertices covering "THE" in the second and the third sequence and connect them with an edge. addEdge(alignG, findVertex(alignG, positionToId(stringSet(alignG),1), 8), findVertex(alignG, positionToId(stringSet(alignG),2), 0)); // Add two vertices covering "FAT" in the second and the third sequence and connect them with an edge. addEdge(alignG, addVertex(alignG, positionToId(stringSet(alignG),1), 11, 3), addVertex(alignG, positionToId(stringSet(alignG),2), 3, 3)); // Add two vertices covering "CAT" in the first and the second sequence and connect them with an edge. addEdge(alignG, addVertex(alignG, positionToId(stringSet(alignG),0), 11, 3), addVertex(alignG, positionToId(stringSet(alignG),1), 14, 3)); // Find the vertex covering "CAT" in the first sequence and add the vertex covering "CAT" in the third sequence and connect them with an edge. addEdge(alignG, findVertex(alignG, positionToId(stringSet(alignG),0), 11), addVertex(alignG, positionToId(stringSet(alignG),2), 6, 3)); // Find the vertices covering "CAT" in the second and the third sequence and connect them with an edge. addEdge(alignG, findVertex(alignG, positionToId(stringSet(alignG),1), 14), findVertex(alignG, positionToId(stringSet(alignG),2), 6)); ::std::cout << alignG << ::std::endl; return 0; }
0 . : . GARFIELDTHE---CAT ||||||||||| ||| GARFIELDTHEBIGCAT ||||||||| --------THEBIGCAT