Seed-and-Extend¶
- Learning Objective
- In this tutorial, you will learn about the seeds-related SeqAn functionality. You will learn how to do seed-and-extend with SeqAn, how to do local and global chaining of seeds. Finally, we will show how to create a banded alignment around a seed chain.
- Difficulty
- Average
- Duration
- 2 h
- Prerequisites
- Sequences
Many efficient heuristics to find high scoring, but inexact, local alignments between two sequences start with small exact (or at least highly similar) segments, so called seeds, and extend or combine them to get larger highly similar regions. Probably the most prominent tool of this kind is BLAST [AGM+90], but there are many other examples like FASTA [Pea90] or LAGAN [BDC+03].
SeqAn’s header file for all data structures and functions related to two-dimensional seeds is <seqan/seeds.h>.
The Seed Class¶

The Seed class allows to store seeds. Seeds have a begin and end position in each sequence. Often, two or more close seeds are combined into a larger seed, possibly causing a shift in horizontal or vertical direction between the begin position of the upper left seed and the end position of the lower right seed. For this reason, the Seed class also stores an upper and a lower diagonal to reflect the expansion between those shifted seeds.
The image to the right shows an example where three smaller seeds (black diagonals) were combined (or “chained locally”) into one larger seed (green nine-sided area). The first seed lies on the begin diagonal, the lowermost seed on the lower diagonal and the uppermost seed on the upper diagonal.
The Simple Seed specialization only stores the begin and end positions of the seed (left-uppermost and right-lowermost corners of green surface) in both sequences and the upper and lower diagonal. The initial diagonals are not stored. The ChainedSeed specialization additionally stores these information. In most cases, the Simple Seed class is sufficient since the best alignment around the seeds has to be determined using a banded alignment algorithm of the seed infixes anyway.
You can get/set the begin and end position in the horizontal/vertical sequences using the functions beginPositionH, beginPositionV, setBeginPositionH, and setBeginPositionV. The band information can be queried and updated using lowerDiagonal, upperDiagonal, setLowerDiagonal, and setUpperDiagonal. Note, we use the capital letters ‘H’ and ‘V’ to indicate horizontal direction and vertical direction, respectively, while the database is always considered as the horizontal sequence and the query as the vertical sequence in the context of sequence alignments.
The following program gives an example of creating seeds as well as setting and reading properties.
// Default-construct seed.
seqan::Seed<seqan::Simple> seed1;
std::cout << "seed1\n"
<< "beginPositionH == " << beginPositionH(seed1) << "\n"
<< "endPositionH == " << endPositionH(seed1) << "\n"
<< "beginPositionV == " << beginPositionV(seed1) << "\n"
<< "endPositionV == " << endPositionV(seed1) << "\n"
<< "lowerDiagonal == " << lowerDiagonal(seed1) << "\n"
<< "upperDiagonal == " << upperDiagonal(seed1) << "\n\n";
// Construct seed with begin and end position in both sequences.
seqan::Seed<seqan::Simple> seed2(3, 10, 7, 14);
setUpperDiagonal(seed2, -7);
setLowerDiagonal(seed2, -9);
std::cout << "seed2\n"
<< "beginPositionH == " << beginPositionH(seed2) << "\n"
<< "endPositionH == " << endPositionH(seed2) << "\n"
<< "beginPositionV == " << beginPositionV(seed2) << "\n"
<< "endPositionV == " << endPositionV(seed2) << "\n"
<< "lowerDiagonal == " << lowerDiagonal(seed2) << "\n"
<< "upperDiagonal == " << upperDiagonal(seed2) << "\n\n";
The output to the console is as follows.
seed1
beginPositionH == 0
endPositionH == 0
beginPositionV == 0
endPositionV == 0
lowerDiagonal == 0
upperDiagonal == 0
seed2
beginPositionH == 3
endPositionH == 7
beginPositionV == 10
endPositionV == 14
lowerDiagonal == -9
upperDiagonal == -7
Assignment 1¶
- Type
- Review
- Objective
- Extend the program above such that seed1 is updated from seed2 and all members (begin positions, end positions, diagonals) are equal to the corresponding member of seed times two. For example, the lower diagonal of seed2 should be 2 * lowerDiagonal(seed1).
- Solution
Seed Extension¶
Seeds are often created quickly using a k-mer index: When a k-mer of a given length is found in both sequences, we can use it as a seed. However, the match can be longer than just k characters. To get longer matches, we use seed extension.
In the most simple case we simply look for matching characters in both sequences to the left and right end of the seed. This is called match extension and available through the extendSeed function using the MatchExtend tag.
// The horizontal and vertical sequence (database and query).
seqan::CharString seqH = "The quick BROWN fox jumped again!";
seqan::CharString seqV = "thick BROWN boxes of brownies!";
// ^^^
// Create seed and print the seeed sequence.
seqan::Seed<seqan::Simple> seed(11, 7, 14, 10);
std::cout << "original\n"
<< "seedH: " << infix(seqH, beginPositionH(seed),
endPositionH(seed)) << "\n"
<< "seedV: " << infix(seqV, beginPositionV(seed),
endPositionV(seed)) << "\n";
// Perform match extension.
seqan::Score<int, seqan::Simple> scoringScheme(1, -1, -1);
extendSeed(seed, seqH, seqV, seqan::EXTEND_BOTH, scoringScheme, 3,
seqan::UnGappedXDrop());
// Print the resulting seed.
std::cout << "result\n"
<< "seedH: " << infix(seqH, beginPositionH(seed),
endPositionH(seed)) << "\n"
<< "seedV: " << infix(seqV, beginPositionV(seed),
endPositionV(seed)) << "\n";
original
seedH: ROW
seedV: ROW
result
seedH: ick BROW
seedV: ick BROW
Assignment 2¶
- Type
- Review
- Objective
- Change the example from above to extend the seed to both sides.
- Solution
#include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/seeds.h> int main() { // The horizontal and vertical sequence (database and query). seqan::CharString seqH = "The quick BROWN fox jumped again!"; seqan::CharString seqV = "thick BROWNIES for me!"; // ^^^ // Create seed and print the seeed sequence. seqan::Seed<seqan::Simple> seed(11, 7, 14, 10); std::cout << "original\n" << "seedH: " << infix(seqH, beginPositionH(seed), endPositionH(seed)) << "\n" << "seedV: " << infix(seqV, beginPositionV(seed), endPositionV(seed)) << "\n"; // Perform match extension. extendSeed(seed, seqH, seqV, seqan::EXTEND_BOTH, seqan::MatchExtend()); // Print the resulting seed. std::cout << "result\n" << "seedH: " << infix(seqH, beginPositionH(seed), endPositionH(seed)) << "\n" << "seedV: " << infix(seqV, beginPositionV(seed), endPositionV(seed)) << "\n"; return 0; }
A more complex case is the standard bioinformatics approach of x-drop extension:
In the ungapped case, we extend the seed by comparing the i-th character to the left/right of the seed of the horizontal sequence with the j-th character to the left/right of the seed in the vertical sequence. Matches and mismatches are assigned with scores (usually matches are assigned with positive scores and mismatches are assigned with negative scores). The scores are summed up. When one or more mismatches occur, the running total will drop. When the sum drops more strongly than a value x, the extension is stopped.
This approach is also available in the gapped case in the SeqAn library. Here, creating gaps is also possible but also assigned negative scores.
// The horizontal and vertical sequence (database and query).
seqan::CharString seqH = "The quick BROWN fox jumped again!";
seqan::CharString seqV = "thick BROWNIES for me!";
// ^^^
// Create seed and print the seeed sequence.
seqan::Seed<seqan::Simple> seed(11, 7, 14, 10);
std::cout << "original\n"
<< "seedH: " << infix(seqH, beginPositionH(seed),
endPositionH(seed)) << "\n"
<< "seedV: " << infix(seqV, beginPositionV(seed),
endPositionV(seed)) << "\n";
// Perform match extension.
extendSeed(seed, seqH, seqV, seqan::EXTEND_LEFT, seqan::MatchExtend());
// Print the resulting seed.
std::cout << "result\n"
<< "seedH: " << infix(seqH, beginPositionH(seed),
endPositionH(seed)) << "\n"
<< "seedV: " << infix(seqV, beginPositionV(seed),
endPositionV(seed)) << "\n";
original
seedH: ROW
seedV: ROW
result
seedH: ick BROWN fox
seedV: ick BROWN box
Assignment 3¶
- Type
- Review
- Objective
- Change the example from above to use gapped instead of ungapped x-drop extension.
- Solution
#include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/score.h> #include <seqan/seeds.h> int main() { // The horizontal and vertical sequence (database and query). seqan::CharString seqH = "The quick BROWN fox jumped again!"; seqan::CharString seqV = "thick BROWN boxes of brownies!"; // ^^^ // Create seed and print the seeed sequence. seqan::Seed<seqan::Simple> seed(11, 7, 14, 10); std::cout << "original\n" << "seedH: " << infix(seqH, beginPositionH(seed), endPositionH(seed)) << "\n" << "seedV: " << infix(seqV, beginPositionV(seed), endPositionV(seed)) << "\n"; // Perform match extension. seqan::Score<int, seqan::Simple> scoringScheme(1, -1, -1); extendSeed(seed, seqH, seqV, seqan::EXTEND_BOTH, scoringScheme, 3, seqan::GappedXDrop()); // Print the resulting seed. std::cout << "result\n" << "seedH: " << infix(seqH, beginPositionH(seed), endPositionH(seed)) << "\n" << "seedV: " << infix(seqV, beginPositionV(seed), endPositionV(seed)) << "\n"; return 0; }
After extending a seed, we might wish to actually get the resulting alignment. When using gapped x-drop extension, we need to perform a banded global alignment of the two sequence infixes that correspond to the seed. This is shown in the following example:
// The horizontal and vertical sequence (database and query).
seqan::CharString seqH = "The quick BROWN fox jumped again!";
seqan::CharString seqV = "thick BROWN boxes of brownies!";
// ^^^
// Create seed and print the seeed sequence.
seqan::Seed<seqan::Simple> seed(11, 7, 14, 10);
// Perform match extension.
seqan::Score<int, seqan::Simple> scoringScheme(1, -1, -1);
extendSeed(seed, seqH, seqV, seqan::EXTEND_BOTH, scoringScheme, 3,
seqan::UnGappedXDrop());
// Perform a banded alignment.
seqan::Align<seqan::CharString> align;
resize(rows(align), 2);
assignSource(row(align, 0), infix(seqH, beginPositionH(seed),
endPositionH(seed)));
assignSource(row(align, 1), infix(seqV, beginPositionV(seed),
endPositionV(seed)));
// TODO(holtgrew): Use seed diagonals as bands.
globalAlignment(align, scoringScheme);
std::cerr << "Resulting alignment\n" << align << "\n";
Resulting alignment
0 . :
ick BROWN fox
|||||||||||||
ick BROWN box
Assignment 4¶
- Type
- Review
- Objective
- Change the example from above to a gap open score of -2 and a gap extension score of -2. Use this scoring scheme for the global alignment as well and thus Gotoh’s algorithm.
- Solution
Note that we do not have to explicitely call Gotoh’s algorithm in globalAlignment(). The fact that the gap extension score is different from the gap opening score is enough.
#include <seqan/align.h> #include <seqan/file.h> #include <seqan/score.h> #include <seqan/seeds.h> #include <seqan/sequence.h> int main() { // The horizontal and vertical sequence (database and query). seqan::CharString seqH = "The quick BROWN fox jumped again!"; seqan::CharString seqV = "thick BROWN boxes of brownies!"; // ^^^ // Create seed and print the seeed sequence. seqan::Seed<seqan::Simple> seed(11, 7, 14, 10); // Perform match extension. seqan::Score<int, seqan::Simple> scoringScheme(1, -1, -1, -2); extendSeed(seed, seqH, seqV, seqan::EXTEND_BOTH, scoringScheme, 3, seqan::GappedXDrop()); // Perform a banded alignment. seqan::Align<seqan::CharString> align; resize(rows(align), 2); assignSource(row(align, 0), infix(seqH, beginPositionH(seed), endPositionH(seed))); assignSource(row(align, 1), infix(seqV, beginPositionV(seed), endPositionV(seed))); // TODO(holtgrew): Use seed diagonals as bands. globalAlignment(align, scoringScheme); std::cerr << "Resulting alignment\n" << align << "\n"; // FRAGMENT(footer) return 0; }
Local Chaining using Seed Sets¶
Usually, we quickly determine a large number of seeds. When a seed is found, we want to find a “close” seed that we found previously and combine it to form a longer seed. This combination is called local chaining. This approach has been pioneered in the CHAOS and BLAT programs.
SeqAn provides the SeedSet class as a data structure to efficiently store seeds and combine new seeds with existing ones. The following example creates a SeedSet object seeds, adds four seeds to it and then prints its contents.
typedef seqan::Seed<seqan::Simple> TSeed;
typedef seqan::SeedSet<seqan::Simple> TSeedSet;
TSeedSet seedSet;
addSeed(seedSet, TSeed(0, 0, 2), seqan::Single());
addSeed(seedSet, TSeed(3, 5, 2), seqan::Single());
addSeed(seedSet, TSeed(4, 2, 3), seqan::Single());
addSeed(seedSet, TSeed(9, 9, 2), seqan::Single());
std::cout << "Resulting seeds.\n";
typedef seqan::Iterator<TSeedSet>::Type TIter;
for (TIter it = begin(seedSet, seqan::Standard()); it != end(seedSet, seqan::Standard()); ++it)
std::cout << "(" << beginPositionH(*it) << ", " << endPositionH(*it)
<< ", " << beginPositionV(*it) << ", " << endPositionV(*it)
<< ", " << lowerDiagonal(*it) << ", " << upperDiagonal(*it)
<< ")\n";
The output of the program above can be seen below.
Resulting seeds.
(3, 5, 5, 7, -2, -2)
(0, 2, 0, 2, 0, 0)
(9, 11, 9, 11, 0, 0)
(4, 7, 2, 5, 2, 2)
Note that we have used the Single() tag for adding the seeds. This forces the seeds to be added independent of the current seed set contents.
By using different overloads of the addSeed, we can use various local chaining strategies when adding seed A.
- Merge
- If there is a seed B that overlaps with A and the difference in diagonals is smaller than a given threshold then A can be merged with B.
- SimpleChain
- If there is a seed B whose distance in both sequences is smaller than a given threshold then A can be chained to B.
- Chaos
- Following the strategy of Chaos [BCGottgens+03], if A is within a certain distance to B in both sequences and the distance in diagonals is smaller than a given threshold then A can be chained to B.
The addSeed function returns a boolean value indicating success in finding a suitable partner for chaining. A call using the Single strategy always yields true.
The following example shows how to use the SimpleChain strategy.
typedef seqan::Seed<seqan::Simple> TSeed;
typedef seqan::SeedSet<seqan::Simple> TSeedSet;
seqan::Dna5String seqH;
seqan::Dna5String seqV;
seqan::Score<int, seqan::Simple> scoringScheme(1, -1, -1);
seqan::String<TSeed> seeds;
appendValue(seeds, TSeed(0, 0, 2));
appendValue(seeds, TSeed(3, 5, 2));
appendValue(seeds, TSeed(4, 2, 3));
appendValue(seeds, TSeed(9, 9, 2));
TSeedSet seedSet;
for (unsigned i = 0; i < length(seeds); ++i)
{
if (!addSeed(seedSet, seeds[i], 2, 2, scoringScheme,
seqH, seqV, seqan::SimpleChain()))
addSeed(seedSet, seeds[i], seqan::Single());
}
std::cout << "Resulting seeds.\n";
typedef seqan::Iterator<TSeedSet>::Type TIter;
for (TIter it = begin(seedSet, seqan::Standard());
it != end(seedSet, seqan::Standard()); ++it)
std::cout << "(" << beginPositionH(*it) << ", " << endPositionH(*it)
<< ", " << beginPositionV(*it) << ", " << endPositionV(*it)
<< ", " << lowerDiagonal(*it) << ", " << upperDiagonal(*it)
<< ")\n";
As we can see, the seed TSeed(4, 2, 3) has been chained to TSeed(0, 0, 2).
Resulting seeds.
(3, 5, 5, 7, -2, -2)
(0, 7, 0, 5, 0, 2)
(9, 11, 9, 11, 0, 0)
Assignment 5¶
- Type
- Review
- Objective
- Change the example above to use the Chaos strategy instead of the SimpleChain.
- Solution
#include <seqan/file.h> #include <seqan/score.h> #include <seqan/seeds.h> #include <seqan/sequence.h> int main() { typedef seqan::Seed<seqan::Simple> TSeed; typedef seqan::SeedSet<seqan::Simple> TSeedSet; seqan::Dna5String seqH; seqan::Dna5String seqV; seqan::Score<int, seqan::Simple> scoringScheme(1, -1, -1); seqan::String<TSeed> seeds; appendValue(seeds, TSeed(0, 0, 2)); appendValue(seeds, TSeed(3, 5, 2)); appendValue(seeds, TSeed(4, 2, 3)); appendValue(seeds, TSeed(9, 9, 2)); TSeedSet seedSet; for (unsigned i = 0; i < length(seeds); ++i) { if (!addSeed(seedSet, seeds[i], 2, 2, scoringScheme, seqH, seqV, seqan::Chaos())) addSeed(seedSet, seeds[i], seqan::Single()); } std::cout << "Resulting seeds.\n"; typedef seqan::Iterator<TSeedSet>::Type TIter; for (TIter it = begin(seedSet, seqan::Standard()); it != end(seedSet, seqan::Standard()); ++it) std::cout << "(" << beginPositionH(*it) << ", " << endPositionH(*it) << ", " << beginPositionV(*it) << ", " << endPositionV(*it) << ", " << lowerDiagonal(*it) << ", " << upperDiagonal(*it) << ")\n"; return 0; }
Global Chaining¶
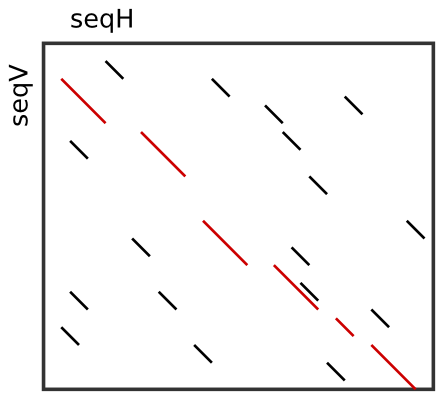
After one has determined a set of candidate seeds, a lot of these seeds will conflict. The image to the right shows an example. Some conflicting seeds might be spurious matches or come from duplication events.
Often, we need to find a linear ordering of the seeds such that each seed starts after all of its predecessor end in both sequences. This can be done efficiently using dynamic sparse programming (in time \(\mathcal{O}(n log n)\) where \(n\) is the number of seeds) as described in [Gus97]. The red seeds in the image to the right show such a valid chain.
This functionality is available in SeqAn using the chainSeedsGlobally function. The function gets a sequence container of Seed objects for the result as its first parameter and a SeedSet as its second parameter. A subset of the seeds from the SeedSet are then selected and stored in the result sequence.
The following shows a simple example.
typedef seqan::Seed<seqan::Simple> TSeed;
typedef seqan::SeedSet<seqan::Simple> TSeedSet;
TSeedSet seedSet;
addSeed(seedSet, TSeed(0, 0, 2), seqan::Single());
addSeed(seedSet, TSeed(3, 5, 2), seqan::Single());
addSeed(seedSet, TSeed(4, 2, 3), seqan::Single());
addSeed(seedSet, TSeed(9, 9, 2), seqan::Single());
seqan::String<TSeed> result;
chainSeedsGlobally(result, seedSet, seqan::SparseChaining());
Assignment 6¶
- Type
- Review
- Objective
- Change the example from above to use a different chain of seeds. The seeds should be TSeed(1, 1, 3), TSeed(6, 9, 2), TSeed(10, 13, 3), and TSeed(20, 22, 5).
- Solution
#include <seqan/sequence.h> #include <seqan/file.h> #include <seqan/seeds.h> int main() { typedef seqan::Seed<seqan::Simple> TSeed; typedef seqan::SeedSet<seqan::Simple> TSeedSet; TSeedSet seedSet; addSeed(seedSet, TSeed(1, 1, 3), seqan::Single()); addSeed(seedSet, TSeed(6, 9, 2), seqan::Single()); addSeed(seedSet, TSeed(10, 13, 3), seqan::Single()); addSeed(seedSet, TSeed(20, 22, 5), seqan::Single()); seqan::String<TSeed> result; chainSeedsGlobally(result, seedSet, seqan::SparseChaining()); return 0; }
Banded Chain Alignment¶
After obtaining such a valid seed chain, we would like to obtain an alignment along the chain. For this, we can use the so-called banded chain alignment algorithm (introduced by Brudno’s LAGAN). Around seeds, we can use banded DP alignment and the spaces between seeds can be aligned using standard DP programming alignment.
In SeqAn you can compute the banded chain alignment by calling the function bandedChainAlignment. This function gets the structure in which the alignment should be stored as the first parameter. This corresponds to the interface of the globalAlignment and allows the same input types. Additionally, this function requires a non-empty, non-decreasing monotonic chain of seeds which is used as the rough global map for computing the global alignment. The third required parameter is the Score.
Note, that there are a number of optional parameters that can be specified. These include a second Score which, if specified, is used to evaluate the regions between two consecutive seeds differently than the regions around the seeds itself (for which then the first specified score is taken.). As for the global alignment you can use the AlignConfig to specify the behavior for initial and end gaps. The last optional parameter is the band extension. This parameter specifies to which size the bands around the anchors should be extended. The default value is 15 and conforms the default value in the LAGAN-algorithm [BDC+03].
Important
At the moment the specified value for the band extension must be at least one.
typedef Seed<Simple> TSeed;
Dna5String sequenceH = "CGAATCCATCCCACACA";
Dna5String sequenceV = "GGCGATNNNCATGGCACA";
String<TSeed> seedChain;
appendValue(seedChain, TSeed(0, 2, 5, 6));
appendValue(seedChain, TSeed(6, 9, 9, 12));
appendValue(seedChain, TSeed(11, 14, 17, 16));
Align<Dna5String, ArrayGaps> alignment;
resize(rows(alignment), 2);
assignSource(row(alignment, 0), sequenceH);
assignSource(row(alignment, 1), sequenceV);
Score<int, Simple> scoringScheme(2, -1, -2);
int result = bandedChainAlignment(alignment, seedChain, scoringScheme, 2);
std::cout << "Score: " << result << std::endl;
std::cout << alignment << std::endl;
The output of the example above.
Score: 5
0 . : . :
--CGAAT--CCATCCCACACA
|| || ||||| ||||
GGCG-ATNNNCATGG--CACA
Assignment 7¶
- Type
- Review
- Objective
Change the example form above to use two different scoring schemes. The scoring scheme for the seeds should use the Levenshtein distance and the score for the gap regions should be an affine score with the following values: match = 2, mismatch = -1, gap open = -2, gap extend = -1.
Furthermore, we are looking for a semi-global alignment here the initial and end gaps in the query sequence are free.
- Solution
#include <seqan/sequence.h> #include <seqan/align.h> #include <seqan/score.h> #include <seqan/seeds.h> int main() { typedef seqan::Seed<seqan::Simple> TSeed; seqan::Dna5String sequenceH = "CGAATCCATCCCACACA"; seqan::Dna5String sequenceV = "GGCGATNNNCATGGCACA"; seqan::Score<int, seqan::Simple> scoringSchemeAnchor(0, -1, -1); seqan::Score<int, seqan::Simple> scoringSchemeGap(2, -1, -1, -2); seqan::String<TSeed> seedChain; seqan::appendValue(seedChain, TSeed(0, 2, 5, 6)); seqan::appendValue(seedChain, TSeed(6, 9, 9, 12)); seqan::appendValue(seedChain, TSeed(11, 14, 17, 16)); seqan::Align<seqan::Dna5String, seqan::ArrayGaps> alignment; seqan::resize(seqan::rows(alignment), 2); seqan::assignSource(seqan::row(alignment, 0), sequenceH); seqan::assignSource(seqan::row(alignment, 1), sequenceV); seqan::AlignConfig<true, false, false, true> alignConfig; int result = seqan::bandedChainAlignment(alignment, seedChain, scoringSchemeAnchor, scoringSchemeGap, alignConfig, 2); std::cout << "Score: " << result << std::endl; std::cout << alignment << std::endl; return 0; }